Les médicaments et les régimes que l’on utilise aujourd’hui pour modifier le métabolisme des lipides agissent sur les acides gras ou le cholestérol mais non sur les apoprotéines par modification de leur synthèse ou de leur fixation aux récepteurs.
À impact sur les acides gras
Modifications de l’apport alimentaire
L’apport d’acides gras saturés, c’est-à-dire de graisses d’origine animale comme le beurre et le lard, ou d’acide gras insaturés de type trans, présents dans certaines margarines, tend à augmenter les LDL considérées comme néfastes et à diminuer les HDL considérées comme bénéfiques. Toutefois l’acide stéarique (C18 : 0, saturé) est peu hypercholestérolémiant car il est rapidement transformé en acide oléique (C18 :1), monoinsaturé) par le foie. L’acide palmitique (C16 :0) est peu hypercholestérolémiant ; les acides myristique (C14 :0) et laurique (C12) le sont davantage.
Parmi les acides gras insaturés de type cis, l’acide oléique (C18 :1) présent dans les huiles végétales, réduit les LDL et augmente les HDL. L’acide linoléique (C18 :2) est moins approprié car il est plus facilement peroxydé par les réactions radicalaires. Les acides gras polyinsaturés de type w3 comme l’acide eicosapentaénoïque, EPA, (C20 :5) et l’acide docohexaénoïque, DHA, (C22 :6), présents dans les poissons des mers froides, inhibent l’agrégation plaquettaire mais ont assez peu d’effet sur le cholestérol. L’enrichissement du régime alimentaire en acides gras insaturés n-3 diminue le risque de troubles du rythme cardiaque, d’accidents coronariens et de mort subite. Une solution simple pour enrichir modérément l’alimentation en acide oléique est la consommation d’olives, d’avocats, d’huile d’arachide ou de noix, riches en acides gras insaturés, arginine et magnésium.
Il existe des préparations diététiques à base d’huiles de poissons, riches en acides gras insaturés de type cis w3 (ex : huile MAXÉPA*, dont la commercialisation a été arrêtée en 2008). Mais la meilleure solution est d’adopter un régime alimentaire de type méditerranéen. Le régime dit méditerranéen, riche en fruits, légumes, céréales complètes, oléagineux , huile d’olive, poissons, avec un peu de vin, et relativement pauvre en viandes et produits laitiers, réduit d’environ 20 % la mortalité toutes causes confondues, la mortalité d’origine cardiovasculaire et par cancer.
Signalons qu’un acide gras saturé à quatre atomes de carbone, l’acide butyrique, CH3CH2CH2COOH, présent dans le beurre possède un effet antinéoplasique confirmé par de nombreuses études in vitro et in vivo.
Les aliments qui ont ou sont supposés avoir un effet bénéfique sur la santé sont parfois appelés « alicaments ».
Inhibiteurs de l’absorption intestinale des acides gras
L’absorption digestive des lipides, notamment des triglycérides, ne se fait qu’après leur hydrolyse sous l’effet de la lipase gastrique puis pancréatique en acides gras et monoglycérides qui, eux, sont absorbés.
Si l’on inhibe ces lipases, on réduit considérablement l’absorption digestive des lipides ingérés.
La tétrahydrolipstatine ou orlistat, dérivé hydrogéné de la lipstatine, substance naturelle produite par Streptomyces toxytricini, est un inhibiteur des lipases. Administré par voie orale, il est peu absorbé par le tube digestif et agit dans la lumière intestinale. L’orlistat inhibe les lipases en formant une liaison ester avec la sérine de leur site actif.
L’orlistat inhibe la lipase et diminue l’absorption digestive des lipides qui sont éliminés dans les selles. L’orlistat tend à provoquer une diminution des triglycérides et une perte de poids et il est indiqué dans le traitement de l’obésité.
Ses effets indésirables les plus fréquents sont des troubles digestifs, selles huileuses, suintement anal, gaz.
À impact sur le cholestérol
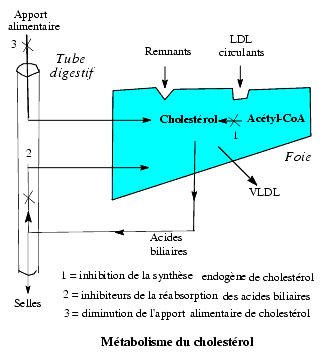
Pour abaisser la concentration de cholestérol dans le plasma, on peut réduire son apport exogène et sa biosynthèse (endogène) ou augmenter son catabolisme.
Réduction de l’apport alimentaire de cholestérol
La réduction de la consommation d’aliments riches en cholestérol, jaune d’uf, abats, beurre, est toujours conseillée mais souvent insuffisante.
Inhibiteurs de l’absorption intestinale du cholestérol, stérols végétaux et ézétimibe
Les stérols et stanols d’origine végétale comme le sitostérol et le sitostanol réduisent l’absorption intestinale de cholestérol et abaissent le LDL-cholestérol plasmatique. Leur effet apparaît d’autant plus important que l’apport alimentaire de cholestérol est élevé. Le sitostanol, présent par exemple dans l’huile de soja, qui lui-même n’est pratiquement pas absorbé est le plus efficace. Cependant il n’existe pas, semble-t-il, d’études démontrant leur efficacité sur la symptomatologie clinique ni sur la mortalité.
Diverses préparations alimentaires, notamment à base de lait écrémé, sont actuellement enrichis en stérols végétaux en vue d’abaisser le taux de cholestérol.
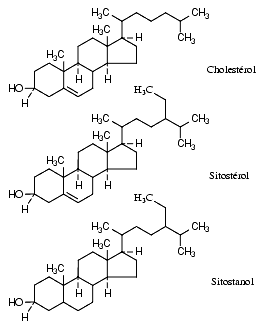
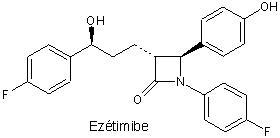
L’ézétimibe est un médicament de synthèse qui inhibe sélectivement l’absorption intestinale de cholestérol et de certains phytostérols comme le sitostérol. Il inhibe le transporteur de stérols. Il agit au niveau de la muqueuse intestinale mais il est lui-même absorbé et partiellement transformé en un métabolite actif glycuronoconjugué, l’ézétimibe-glycuronide. Lors des essais cliniques de courte durée, l’ézétimide a abaissé de près de 20 % la concentration de LDL cholestérol plasmatique.
L’indication de l’ézétimibe, en complément d’une statine, est l’hypercholestérolémie. Seul, sans statine, il a l’indication sitostérolémie, maladie rare due à une absorption intestinale excessive et à une élimination insuffisante de sitostérol, stérol d’origine végétale.
|
Ezétimibe |
EZETROL*, Cp à 10 mg |
|
Ezétimibe + Simvastatine |
INEGY* |
Les études cliniques n’ont pas pour le moment permis de prouver que l’ézétimibe réduit la fréquence des accidents cardiovasculaires ni qu’il retarde la mortalité toutes causes confondues. Elles ne garantissent pas non plus sa bonne tolérance en utilisation prolongée.
Inhibiteurs de la biosynthèse du cholestérol, les statines
Les médicaments actuellement utilisés inhibent l’hydroxy-méthyl-glutaryl-CoA (HMGCoA) réductase, enzyme assurant la transformation du HMG-CoA en mévalonate. Cette réaction constitue l’étape limitante de la synthèse de cholestérol.
Ces inhibiteurs, désignés sous le nom de statines, ont été découverts au Japon vers 1975 dans des cultures de Penicillium; le premier inhibiteur efficace a été la mévastatine. L’inhibition de la HMG-CoA réductase entraîne une diminution très importante de la synthèse endogène de cholestérol, synthèse qui n’est cependant pas totalement supprimée. L’organisme a donc à sa disposition du cholestérol d’origine exogène et, en partie, d’origine endogène. L’activité de la HMG-CoA réductase est plus importante la nuit que le jour et les inhibiteurs sont pris de préférence le soir au coucher.

La diminution de la synthèse endogène de cholestérol, provoquée par les inhibiteurs de la HMG-CoA réductase, induit une augmentation de la synthèse de HMG-CoA réductase qui est ensuite inhibée et surtout une augmentation des récepteurs hépatiques aux LDL, fait confirmé par l’augmentation de l’ARN messager correspondant. Cette inhibition induit en conséquence une captation hépatique majorée des LDL dont la concentration plasmatique s’abaisse.
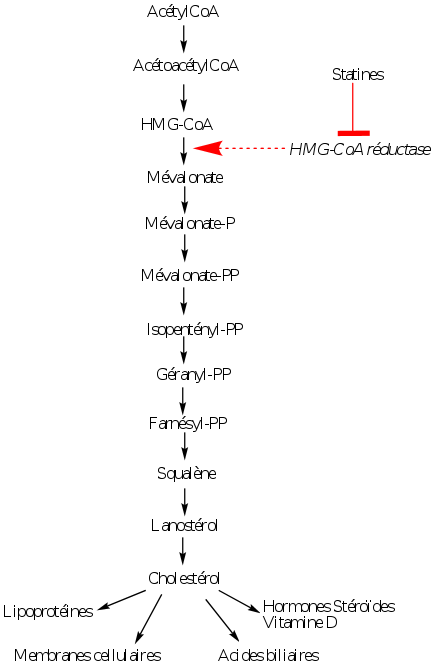
Par ce mécanisme les inhibiteurs de la HMG-CoA réductase entraînent un abaissement dose-dépendant du cholestérol de type LDL allant de 20 à 50%. Il y a également une diminution des triglycérides mais de moindre importance, de 15 à 30%.
L’abaissement de la concentration des lipides sanguins ne constitue pas une preuve de l’efficacité de ces médicaments, leur but est de réduire la fréquence et la gravité des accidents cardiovasculaires d’origine athéromateuse et de diminuer la mortalité. Il est démontré qu’un traitement par la simvastatine ou la pravastatine diminue la concentration plasmatique des LDL et la fréquence des accidents coronaires, infarctus du myocarde, des pontages, des angioplasties avec réduction de la mortalité par coronaropathies et de la mortalité globale, toutes causes confondues. Ces résultats favorables ont été obtenus en prévention primaire et secondaire.
La diminution de la mortalité par les inhibiteurs de la HMG-CoA réductase, qui est le but à atteindre, ne démontre pas nécessairement que c’est l’abaissement du cholestérol plasmatique lui-même qui en est la seule cause. En effet les inhibiteurs de la HMG-CoA réductase diminuent aussi la concentration des précurseurs du cholestérol comme le farnesylpyrophosphate et le géranylpyrophosphate, qui interviennent dans divers processus cellulaires, notamment la farnélysation de Ras, condition nécessaire à son activation et à celle de la voie de signalisation MAPK. La pravastatine s’est montrée efficace dans la prévention des accidents cardiovasculaires chez des coronariens ayant une cholestérolémie normale et a réduit leur mortalité totale.
Les statines utilisées en France, outre les plus anciens, la simvastatine et la pravastatine, sont la fluvastatine, l’atorvastatine et la rosuvastatine.
|
VASTEN* Comprimés à 10, 20 et 40 mg |
|
|
LESCoL* Comprimés à 20, 40 et 80 mg |
|
| Rosuvastatine | CRESTOR*, Comprimés à 5, 10 et 20 mg |
La cérivastatine a été commercialisée dans certains pays puis retirée du commerce en raison d’effets indésirables.

Les principaux effets indésirables des inhibiteurs de l’HMG-CoA réductase, généralement dose-dépendants, sont d’ordre hépatique et musculaire :
- Troubles hépatiques se limitant à une élévation des transaminases qu’il est nécessaire de surveiller au cours du traitement
- Douleurs et faiblesse musculaires qui doivent conduire au dosage de la créatine phosphokinase et à l’arrêt de leur prescription et très rarement des polyneuropathies
- Diminution de la synthèse d’ubiquinone (coenzyme Q10) sans que ses conséquences cliniques aient été démontrées.
D’autres effets indésirables, de type digestif par exemple, ont été également signalés.
Par ailleurs, la simvastatine est métabolisée par le cytochrome P450 3A4; lorsque ce dernier est inhibé par d’autres médicaments comme l’itraconazole ou le jus de pamplemousse, sa concentration plasmatique s’élève considérablement. L’atorvastatine est aussi métabolisée par le cytochrome P450 3A4 et des interactions sont possibles, surtout lorsqu’elle est utilisée à doses très élevées.
Ces inhibiteurs sont contre-indiqués au cours de la grossesse mais la simvastatine, lorsqu’elle a été prise par des femmes enceintes ne semble pas avoir augmenté le risque tératogène.
Le tiadénol a une structure chimique différente de celle des autres hypolipémiants. Il abaisse les lipides sanguins par des mécanismes complexes et mal élucidés, probablement une diminution de la synthèse de cholestérol par inhibition des premières étapes avant le mévalonate. Jusqu’à présent, peu d’effets indésirables ont été signalés lors de sa prescription.
Activateurs indirects du catabolisme du cholestérol, cholestyramine
Le principal médicament de ce groupe est la cholestyramine. C’est une résine échangeuse d’anions qui abaisse indirectement le cholestérol sanguin.
La cholestyramine est un polymère de poids moléculaire élevé, comportant des groupes ammonium quaternaire. Administrée sous forme de chlorure, elle échange les anions chlorure contre d’autres anions, en particulier les acides biliaires.
La cholestyramine n’est pas absorbée par le tube digestif. Son effet est seulement intestinal : elle fixe les acides et en particulier les acides biliaires qui sont éliminés dans les selles, liés à elle. En supprimant le cycle entéro-hépatique des acides biliaires, elle diminue la concentration d’acides biliaires dans les cellules hépatiques, ce qui entraîne une activation de la 7-a-hydroxylase, enzyme responsable de la transformation du cholestérol en acides biliaires. En accélérant le catabolisme du cholestérol, elle provoque indirectement une augmentation des récepteurs LDL au niveau hépatique et donc une captation accélérée des LDL circulants et une chute du cholestérol.
La cholestyramine entraîne, au bout d’une dizaine de jours de traitement, une chute du cholestérol d’environ 20% par abaissement des LDL. Cet abaissement est accompagné d’une élévation – au moins transitoire – des triglycérides et la cholestyramine ne doit pas être utilisée en cas d’hypertriglycéridémie.
L’indication de la cholestyramine est l’hypercholestérolémie isolée (par augmentation des LDL). Elle a été également utilisée pour traiter le prurit provoqué par l’accumulation d’acides biliaires dans le sang des malades ayant une cholestase.
L’association cholestyramine et inhibiteur de l’HMG-CoA réductase est d’une très grande efficacité pour faire chuter le cholestérol en cas d’hypercholestérolémie majeure.
L’effet indésirable le plus fréquent est la constipation avec flatulence et stéatorrhée. On peut aussi observer une hyperchlorémie car la résine libère des ions chlorure qui sont absorbés par le tube digestif.
En raison de sa non-spécificité vis-à-vis des acides biliaires, la cholestyramine fixe et empêche l’absorption intestinale de tous les médicaments à fonction acide, diurétiques, antivitamines K, anti-inflammatoires non stéroïdiens et des vitamines A, D, E, K, dont l’absorption est favorisée par les sels biliaires.
En principe, la cholestyramine ne pas doit être prise en même temps que les autres médicaments mais généralement 2 à 4 heures après.
Un inconvénient de la cholestyramine est qu’il en faut une quantité importante pour obtenir un effet et la prise trois fois par jour d’un paquet de poudre à mettre en suspension dans un verre d’eau peut être rebutante.
« Elévateurs » du HDL-cholestérol
Le HDL-cholestérol ayant un effet anti-athérogène, on a cherché à augmenter sa concentration plasmatique. Les moyens qui sont connus pour augmenter modérément sa concentration sont une consommation modérée d’alcool et la pratique de l’exercice physique. Parmi les médicaments qui élèvent le plus la concentration le HDL-cholestérol, on peut citer l’acide nicotinique.
La CETP, ou cholesteryl ester transfer protein, est une glycoprotéine présente dans le plasma qui facilite le transfert des esters du cholestérol du HDL (apolipoprotéine A-1) vers les VLDL et les LDL (apolipoprotéine-B). L’inhibition de la CETP tend à augmenter la concentration de HDL-cholestérol.
Le torcetrapib est un inhibiteur de la CETP, il augmente la concentration de HDL-cholestérol et diminue celle des LDL, mais au cours des essais cliniques il a augmenté la mortalité et son étude a été abandonnée, sans qu’on sache s’il s’agit d’un effet liée à la molécule elle-même ou à l’inhibition de la CETP.
Hypocholestérolémiants à mécanismes d’action complexe
Fibrates
Le premier médicament de ce groupe, le clofibrate, dérivé de l’acide aryl-oxy-butyrique, a été commercialisé à partir de 1965. D’autres dérivés dont la structure chimique présente une très grande ressemblance avec celle du clofibrate ont été commercialisés ultérieurement : le gemfibrozil, le fénofibrate, le ciprofibrate et le bézafibrate qui sont appelés « fibrates de deuxième génération ».
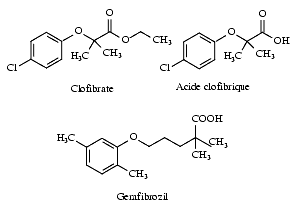
Bien qu’il s’agisse de médicaments utilisés depuis de nombreuses années, leur mécanisme d’action est mal connu. Leur effet principal serait dû à l’activation de la lipoprotéine lipase conduisant à une hydrolyse accélérée des VLDL ainsi que des LDL. Une inhibition de l’HMG-CoA réductase a été évoquée, mais elle n’est guère compatible avec les résultats cliniques.
L’acide clofibrique et ses dérivés, chez le rat mais apparemment pas chez l’homme, activent les PPAR a et b (peroxisome proliferator activated receptor) qui alors forment des hétérodimères avec les récepteurs de l’acide rétinoïque. Ces hétérodimères fonctionnent comme facteurs transcriptionnels favorisant notamment la formation de peroxisomes. Les peroxisomes, présents notamment dans les hépatocytes, sont des vésicules contenant des enzymes qui, en présence d’oxygène moléculaire, assurent l’oxydation de substrats avec formation de peroxyde d’hydrogène. Les conséquences bénéfiques ou néfastes de cette action n’ont pas été clairement précisées.
Le clofibrate et ses analogues abaissent peu le cholestérol sanguin mais font chuter nettement les triglycérides en agissant sur les VLDL. Administrés à des malades porteurs d’une élévation des VLDL, ils ont pu entraîner une régression des xanthomes (dépôts lipidiques non vasculaires) mais les triglycérides ne semblent pas jouer un rôle important dans le développement de l’athérome.
Administrés sous forme d’acides ou d’esters, les fibrates sont bien absorbés par le tube digestif. Les esters sont hydrolysés en acides, seule forme active, par exemple le clofibrate est hydrolysé en acide clofibrique. Les acides se lient aux protéines plasmatiques. Ils peuvent déplacer les antivitamines K liés aux protéines plasmatiques.
Le produit le mieux connu de ce groupe est le clofibrate. Dans les études cliniques contre groupe témoin, le clofibrate réduit la fréquence des infarctus du myocarde d’environ 20%, mais il double celle des lithiases biliaires et a entraîné une surmortalité, d’origine non précisée. Il n’est plus commercialisé.
Les fibrates en général, bien que n’ayant pas fait la preuve de leur efficacité en réduisant la mortalité globale au cours d’études comparatives, ont été très largement prescrits. Dans une étude, le gemfibrozil a réduit la mortalité d’origine cardiovasculaire sans toutefois réduire la mortalité toutes causes confondues.
Les effets indésirables les plus fréquents du clofibrate, et probablement des autres dérivés, sont digestifs et musculaires.
- digestifs : dyspepsie, flatulence, augmentation des transaminases, hépatomégalie, lithiase biliaire.
- musculaires : crampes, faiblesse musculaire, myosite, myalgie, élévation de la créatine phosphokinase, exceptionnellement rhabdomyolyse.
- divers : fatigue, prise de poids, alopécie…
Il est à noter que les fibrates potentialisent l’effet des anticoagulants oraux, par compétition de fixation sur les protéines plasmatiques et par altération de la synthèse des facteurs vitamine K-dépendants.
Ils sont contre-indiqués pendant la grossesse.
Acide nicotinique
L’acide nicotinique, connu depuis plus de 50 ans, a été utilisé en thérapeutique, à faible dose de l’ordre du 1 mg comme vasodilatateur et à fortes doses, de l’ordre du gramme, comme hypolipémiant ; il possède de plus des propriétés de type vitamine PP après sa transformation dans l’organisme en nicotinamide qui théoriquement ne donne pas de vasodilatation.
L’acide nicotinique a des effets bénéfiques sur les lipides sanguins : il abaisse le LDL-cholestérol, augmente le HDL-cholestérol, diminue Lp(a), abaisse les triglycérides, par des mécanismes complexes. Cependant aucune étude clinique ne semble avoir démontré que l’acide nicotinique diminuait le risque d’accidents cardiovasculaires ni la mortalité toutes causes confondues.
La commercialisation de l’acide nicotinique (préparations à libération non retardée) a été interrompue pendant plusieurs années en France ; une préparation à libération prolongée, Niaspan LP*, a été commercialisée. Cette libération prolongée diminue l’intensité des bouffées de chaleur communément observée avec l’acide nicotinique mais ne réduit pas ses autres effets indésirables : dyspepsie, nausées, vomissements, diarrhée, hépatotoxicité, aggravation du diabète, troubles oculaires : amblyopie toxique et oedème maculaire Par ailleurs, l’association d’acide nicotinique à une statine semble augmenter le risque d’atteinte musculaire et de rhabdomyolyse.
|
NIASPAN* LP, Cp à 375, 500, 750 et 1000 mg |
La vasodilatation provoquée par l’acide nicotinique résulterait de la stimulation des récepteurs PD2 de la prostaglandine PGD2. Le laropiprant est un produit de synthèse en cours d’expérimentation qui inhibe les récepteurs PD2 et diminue ainsi les flushs provoqués par l’acide nicotinique.
