Chaque effet est caractérisé par son intensité et sa durée.
Effet en fonction de la dose
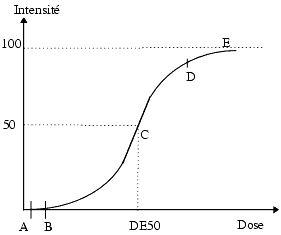
L’effet bénéfique ou toxique d’un principe actif a une intensité qui dépend de la dose administrée et par conséquent de sa concentration plasmatique ou tissulaire. Si l’on trace une courbe effet/dose, la dose étant exprimée en échelle logarithmique, on obtient une courbe sigmoïde qui montre que jusqu’à une certaine dose aucun effet n’apparaît (AB) puis que l’effet augmente avec la dose (BD) jusqu’à atteindre un effet maximum ou plateau (E). La dose qui donne 50% de l’effet maximum s’appelle la dose efficace 50 (DE 50).
Les accidents allergiques ou anaphylactiques sont dits non dose-dépendants. L’expression « non dose-dépendant » signifie que chez une personne sensibilisée à une substance un accident peut survenir lors de l’administration d’une quantité minime de cette substance, par exemple un choc anaphylactique au tout début de la perfusion intraveineuse d’un médicament auquel le malade est sensibilisé. En réalité, ces accidents sont aussi dose-dépendants, mais surviennent chez certains sujets à des doses considérées comme normales, voire extrêmement faibles. On sait en effet que l’on peut réduire la dose d’une substance de telle manière que même les sujets sensibles n’y réagissent plus: c’est le principe des explorations allergologiques où l’on commence par des doses extrêmement faibles.
Le traitement homéopathique (Hahnemann, 1755-1843) apparaît comme une exception à cette règle générale que pour agir un médicament doit être présent dans l’organisme. En effet, d’après la loi des dilutions infinitésimales, les remèdes homéopathiques sont efficaces à des dilutions « infinies » où, théoriquement, ils ne contiennent plus de molécule provenant de la substance initiale. Les homéopathes admettent que la substance de départ a transmis certaines propriétés particulières au solvant qui est dit « dynamisé ». Par exemple, une préparation 15 CH (15ième Centésimale Hahnemannienne) correspond à un facteur de dilution de 1/1030 et ne doit plus guère contenir de molécules de la substance de départ.
Différents types de courbes effet/dose
Selon la nature des principes actifs, on obtient diverses courbes effet/dose. La figure 2 représente les courbes de trois médicaments différents M1, M2 et M3
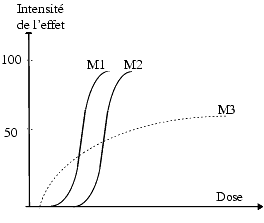
- M1 est plus efficace que M2, c’est-à-dire agit à doses plus faibles;
- M2 est moins efficace que M1 mais en augmentant la dose, on peut obtenir un effet maximum identique.
- Lorsque deux médicaments M1 et M2 donnent des courbes parallèles, on peut penser qu’ils agissent par un mécanisme similaire. La preuve de l’existence d’un mécanisme d’action similaire peut être apportée par l’emploi d’antagonistes spécifiques.
- Le choix entre M1 et M2 dépendra du rapport efficacité/ tolérance de chacun. M1 sera utilisé à doses plus faibles que M2 pour obtenir le même effet mais si M2 est mieux toléré, il peut être préféré à M1.
- M3 commence à agir à faible dose mais l’effet augmente peu lorsque la dose augmente et l’effet maximum obtenu est plus faible que celui de M1 et M2.
Il est évident que ce type de courbes est obtenu chez l’animal ou sur des organes isolés car chez l’homme il est, le plus souvent, difficile d’atteindre sans risque la dose donnant l’effet maximum et de répéter les administrations afin d’obtenir les points nécessaires pour tracer les courbes.
Courbes effet/dose et antagonistes
On utilise les courbes effet/dose pour la recherche de molécules antagonistes ou inhibitrices. On appelle antagoniste toute substance qui diminue ou supprime un ou plusieurs effets d’une autre molécule. En présence de l’antagoniste, la courbe effet/dose est soit simplement décalée soit modifiée.
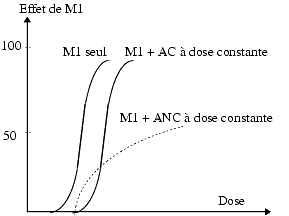
Dans ce type d’expérience, l’antagoniste est présent à une concentration fixe. On augmente progressivement les concentrations de l’agoniste M1 et on constate soit un décalage de la courbe qui reste parallèle à celle de M1 – on dit qu’il s’agit d’un antagoniste compétitif, AC – soit une modification de la courbe qui n’est plus parallèle à A – on dit qu’il s’agit d’un antagoniste non compétitif, ANC. Dans les deux cas, en présence d’un antagoniste, il faut des doses plus élevées de M1 pour atteindre l’effet obtenu en son absence.
En pratique on ne se contente pas de cet aspect qualitatif pour préciser le type d’effet inhibiteur d’un médicament, on utilise des méthodes quantitatives dérivant de celles qui sont employées en enzymologie.
Effets multiples d’un même médicament
Assez souvent, la même molécule possède, à côté de son effet principal, des effets accessoires souvent indésirables. Par exemple, un antihistaminique peut être sédatif ou atropinique; les antidépresseurs tricycliques sont pour la plupart atropiniques, ce qui n’est pas nécessaire à l’effet antidépresseur mais entraîne des effets indésirables comme une sécheresse de la bouche, une constipation, etc.
Lorsque le même médicament a deux ou plusieurs effets différents, ceux-ci peuvent être obtenus à des concentrations tissulaires identiques ou différentes.
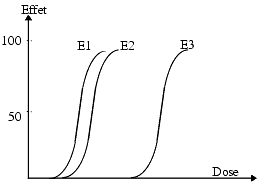
Si l’on admet qu’un médicament M a des effets E1 et E2: si E1 est un effet recherché et E2 un effet indésirable, tout effet bénéfique de E1 sera accompagné de l’effet indésirable E2, car l’un et l’autre sont obtenus à des concentrations presque identiques. C’est le cas de la plupart des anticancéreux et, faute de mieux, on accepte le compromis.
Si l’on admet que le médicament M a un effet E1 bénéfique et un effet E3 indésirable, l’effet bénéfique E1 ne doit pas être accompagné d’effet indésirable E3, car celui-ci n’apparaît qu’à doses très élevées. C’est par exemple le cas du paracétamol qui ne donne une atteinte hépatique grave qu’à des doses conduisant à des concentrations très supérieures à celles que l’on observe lorsqu’il est pris à posologie normale.
Effet en fonction du temps
Début de l’effet
Fréquemment, le délai d’apparition de l’effet dépend de la voie d’administration. Par exemple, un antagoniste adrénergique a1 donne une chute immédiate de la pression artérielle après administration intraveineuse, une chute retardée après administration orale. Ce délai est d’origine pharmacocinétique et correspond au temps nécessaire au médicament pour atteindre ses cibles.
Parfois l’effet apparent est retardé quelle que soit la voie d’administration. En général, dans ce cas, le délai n’est pas d’origine pharmacocinétique, le médicament a atteint sa cible mais l’effet n’apparaît pas pour autant immédiatement. C’est le cas des antidépresseurs tricycliques : même en administration intraveineuse, l’effet antidépresseur n’apparaît qu’en une à trois semaines et ceci à condition de l’administrer quotidiennement pendant l’intervalle
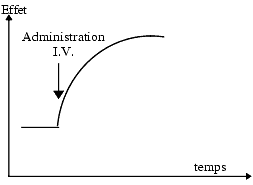
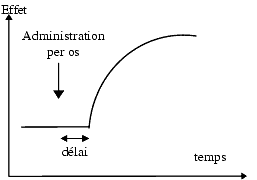
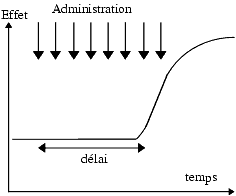
Durée de l’effet
En règle générale, l’effet d’un médicament persiste tant que le médicament ou son métabolite actif est présent dans l’organisme à une concentration suffisante.
Dans certains cas, l’effet du médicament persiste alors que lui-même ou son métabolite actif ne sont plus détectés dans le sang mais sont présents au niveau de certains sites. Il s’agit le plus souvent de médicaments qui inhibent irréversiblement certaines enzymes et dont l’effet dure jusqu’au renouvellement de l’enzyme. C’est le cas d’inhibiteurs de la pompe à protons comme l’oméprazole, de certains inhibiteurs de la monoamine oxydase. C’est aussi le cas de l’aspirine qui inhibe les cyclooxygénases plaquettaires par acétylation irréversible; il faut attendre le renouvellement des plaquettes pour retrouver l’activité initiale.
En règle générale, on cherche à obtenir des médicaments ayant une longue durée d’action – éventuellement par des formes à libération prolongée – pour réduire la fréquence de leur administration. Toutefois, ce qui est habituellement un avantage peut devenir un inconvénient lorsque le médicament considéré est à l’origine d’un effet indésirable grave car il n’est plus possible, en urgence, de soustraire l’organisme à son effet. Ainsi, avant de prescrire un médicament à très longue durée d’action il faut envisager ce que l’on pourrait faire s’il était à l’origine d’un effet indésirable grave.
Effets observés à l’arrêt ou à l’élimination du médicament
- Lorsqu’un médicament est arrêté dans de bonne conditions, après guérison, les effets liés à son arrêt sont négligeables ou imperceptibles.
- Lorsqu’un médicament efficace est arrêté trop tôt, il y a poursuite ou aggravation de la maladie traitée.
- Lorsque un médicament même efficace, à l’origine d’un effet indésirable plus grave que la maladie elle-même, est arrêté, on observe une amélioration de l’état du malade.
- Lorsque l’arrêt d’un médicament entraîne une exacerbation de certains symptômes, on parle de phénomène de rebond. On peut observer à l’arrêt de certains médicaments antihypertenseurs une élévation de la tension artérielle à des valeurs plus élevées qu’avant traitement.
- Lorsque l’arrêt d’un médicament comme la morphine, ou le plus souvent d’une drogue comme la cocaïne, entraîne un besoin impérieux, voir irrépressible, incontrôlable, de le reprendre, on parle de dépendance ou addiction. On distingue schématiquement :
- une dépendance psychique, appelée aussi assuétude, qui est le désir ou le besoin impérieux de continuer à prendre le médicament ou la drogue pour retrouver les sensations qu’il donne.
- une dépendance physique ou syndrome de sevrage ou d’abstinence, caractérisée par l’apparition de symptômes physiques parfois extrêmement marqués et généralement opposés à ceux que donnait le produit arrêté. Ces réactions s’expliquent par la rupture de équilibre établi: l’organisme s’adapte en s’opposant à certains effets du médicament dont l’arrêt brutal crée un déséquilibre transitoire, en sens opposé. Ainsi, l’arrêt brutal d’un médicament sédatif peut entraîner un état d’excitation transitoire, voire même des convulsions. La morphine donne un myosis, son arrêt après utilisation prolongée et à doses élevées donne une mydriase.
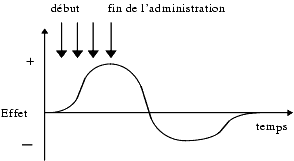
- L’intensité de l’addiction, dépendance psychique et physique, dépend du produit utilisé et est d’autant plus grande que la quantité prise est élevée, que la durée de consommation s’est prolongée et que la demi-vie du produit est courte car c’est la diminution rapide de sa concentration dans l’organisme qui est à l’origine de l’état dysphorique.
- La reprise du produit fait disparaître l’état de besoin et les symptômes physiques. Lorsque la prise d’un produit B chimiquement différent d’un produit A à l’origine de la dépendance, supprime l’état de besoin on parle de dépendance croisée. L’utilisation de la méthadone à la place de la morphine en est un exemple.
- L’administration d’un antagoniste de la substance à l’origine de la dépendance, comme la naloxone à un morphinomane, entraîne un état de manque immédiat.
- Les mécanismes à l’origine de la dépendance physique et psychique sont complexes et apparemment différents.
- Les substances provoquant une dépendance physique agiraient au niveau du Locus ceruleus en inhibant l’adénylcyclase, ce qui diminuerait la concentration d’adénosine monophosphate cyclique (AMPc). À l’arrêt brutal de la prise, c’est-à-dire à la levée de l’inhibition, les mécanismes compensateurs l’emportent, entraînant une hyperstimulation des neurones du Locus ceruleus.
- Les substances entraînant une dépendance psychique agiraient sur le système dopaminergique mésolimbique qui se projette sur le Nucleus accumbens. La dopamine joue un rôle dominant dans les mécanismes de la récompense.
- L’apparition d’un état de dépendance dépend des individus: certains présentent une vulnérabilité particulière et peuvent devenir dépendants dès la ou les premières prises d’une drogue alors que d’autres ne le deviendront pas ou même éprouveront une réaction d’aversion à son égard. Cette susceptibilité particulière dépend de facteurs génétiques non encore déterminés et de facteurs environnementaux. Le stress, sans doute par l’intermédiaire de la libération de glucocorticoïdes, favorise l’installation d’un état de dépendance vis-à vis des drogues.
- A la notion de dépendance il faut opposer la notion de répulsion ou d’aversion, beaucoup moins bien connue que la précédente. Si on offre à un animal le choix entre une boisson contenant de la chlorpromazine et de l’eau distillée, il préférera l’eau distillée. Par contre, si on lui présente une boisson à base de cocaïne, il en consommera jusqu’à en mourir (dépendance).
Variation de l’effet après administrations répétées
L’effet d’un médicament M1 administré d’une manière répétée à la même dose et dans les mêmes conditions peut rester inchangé, diminuer ou augmenter.
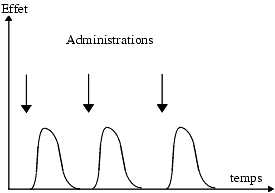
Effet inchangé
C’est le cas le plus fréquent : l’effet du médicament reste identique lorsqu’il est administré d’une façon répétée.
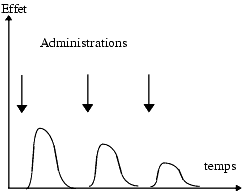
Effet diminué
Lorsque l’effet obtenu décroît progressivement au cours d’administrations successives et rapprochées, on dit qu’il y a tachyphylaxie. La tachyphylaxie évoque la libération et l’épuisement progressif des réserves d’un produit endogène actif, libéré sous l’effet du médicament. C’est le cas, par exemple, de l’éphédrine qui libère des catécholamines.
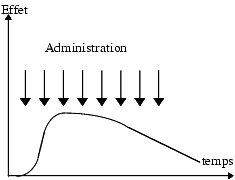
Lorsque l’effet obtenu décroît au cours d’une administration chronique, on parle de tolérance ou encore d’accoutumance, par exemple tolérance à la morphine chez le morphinomane qui utilise des doses de plus en plus élevées pour compenser la perte de son efficacité. Il faut remarquer que le mot tolérance peut être utilisé dans un sens différent : celui d’absence d’effet néfaste et on parle de bonne tolérance, de bien toléré.
Effet augmenté
Ce cas est assez exceptionnel et résulte en général de l’accumulation du médicament dans l’organisme en raison par exemple de l’apparition d’une saturation d’un processus d’inactivation.
Rapport efficacité/toxicité en fonction de la dose
Comme, d’une manière générale, l’augmentation des doses augmente à la fois l’efficacité et la toxicité, il faut tenir compte du rapport efficacité/toxicité en fonction de la dose. Cette courbe montre qu’il existe une dose optimum. En pharmacologie clinique, il est difficile de tracer de telles courbes, mais le souci de prescrire la dose minimum efficace doit être toujours présent à l’esprit du médecin.
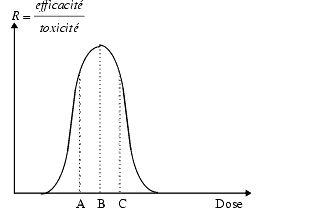
La figure ci-dessus montre qu’à faible dose, inférieure à A, le médicament est peu efficace et qu’à une dose supérieure à C il devient toxique. L’optimum se situe entre A et C.
Différences interindividuelles
On considère que les effets des médicaments – qu’ils soient bénéfiques ou indésirables – sont globalement semblables lorsqu’ils sont prescrits à la même posologie et dans les mêmes circonstances à des individus ayant les mêmes caractéristiques, sexe, poids, âge etc.
En réalité il existe des différences de réponses aux médicaments selon les individus; lorsque ces différences sont faibles, on les néglige; lorsqu’elles sont suffisamment importantes pour avoir des conséquences décelables chez les malades, on doit les prendre en compte.
Les différences interindividuelles sont soit d’origine pharmacocinétique et sont liées à des différences de métabolisme des médicaments (absorption, biotransformations, élimination) conduisant à des concentrations différentes au niveau des cibles, soit d’origine pharmacodynamique se manifestant par une réponse différente des cibles à des concentrations similaires.
Ces différences interindividuelles reposent généralement sur des différences de structure et d’activité de protéines impliquées soit dans le métabolisme du médicament, soit dans l’activité de ses cibles. Ces différences peuvent être soit acquises et sont de type environnemental (maladie, vieillissement, alcoolisme, tabagisme, toxicomanie etc…) soit, le plus souvent, constitutives de l’individu et sont d’origine génétique et transmissibles à la descendance. La comparaison du métabolisme et des effets des médicaments chez des jumeaux vrais et des faux jumeaux permet de différencier l’influence des facteurs environnementaux de celle des facteurs génétiques. La partie de la pharmacologie qui étudie ce dernier type de différences s’appelle la pharmacogénétique. L’étude de la transmission héréditaire de ces particularités dépasse le cadre de cet ouvrage.
Il existe de nombreux exemples de différences de type pharmacocinétique d’origine génétique comme l’hydrolyse de la succinylcholine par les pseudocholinestérases, l’acétylation de l’isoniazide par les N-acétyltransférases, l’oxydation de divers médicament par les cytochromes P-450.
Parmi les différences d’origine pharmacodynamique, on peut citer la résistance à l’insuline, la sensibilité aux effets arythmogènes de certains médicaments chez les personnes présentant le syndrome du QT long à l’électrocardiogramme.
La connaissance de ces différences, qu’elles soient acquises ou constitutives, de type pharmacocinétique ou pharmacodynamique, et en pratique ceci est généralement intriqué, peut être utile pour choisir un médicament ou adapter sa posologie.
