Les médicaments réduisant l’hyperglycémie peuvent être classés en trois groupes :
- les insulomimétiques directs qui activent les récepteurs de l’insuline
- les insulinomimétiques indirects qui augmentent la libération d’insuline comme les sulfamides hypoglycémiants ou potentialisent l’effet de l’insuline comme la metformine
- les médicaments qui agissent directement sur le métabolisme du glucose comme les inhibiteurs des a-glucosidases et les inhibiteurs de l’aldose réductase.
Insulomimétiques directs ou agonistes
Il n’y a pas pour le moment de médicament disponible autre que l’insuline elle-même à agir directement sur ses récepteurs.
De nombreuses études sont en cours pour trouver des médicaments non peptidiques susceptibles d’activer le récepteur de l’insuline en agissant soit sur la partie extramembranaire soit sur la partie intracytoplasmique du récepteur de l’insuline.
Le vanadium, sous forme de vanadate, à doses pharmacologiques, a un effet insulinomimétique direct et indirect, mais il n’est pas commercialisé comme médicament. Son intérêt thérapeutique qui semble réel reste à confirmer.
Activateurs de la sécrétion d’insuline
Les premiers activateurs de la sécrétion d’insuline à être connus ont été les sulfamides hypoglycémiants.
Sulfamides hypoglycémiants
L’effet hypoglycémiant des sulfamides a été découvert à Montpellier vers 1945 par Janbon qui observa l’effet hypoglycémiant d’un sulfamide utilisé comme antibactérien, et par Loubatière qui en précisa le mécanisme d’action en montrant que l’effet hypoglycémiant résultait d’une augmentation de la sécrétion d’insuline par le pancréas.
Effets
Les sulfamides hypoglycémiants stimulent la sécrétion d’insuline par les cellules ß du pancréas en les sensibilisant à l’action du glucose.
Ils se lient à un récepteur situé sur la membrane plasmique, appelé SUR (sulfonylurea receptor), et dont on ne connaît pas le médiateur endogène, et inhibent l’efflux de potassium de la cellule ß par fermeture des canaux potassiques ATP-dépendants. L’élévation de la concentration de potassium intra-cellulaire qui s’ensuit crée une dépolarisation cellulaire suffisante pour déclencher l’ouverture des canaux calcium voltage-dépendants. C’est en définitive l’augmentation du Ca2+ intracellulaire qui provoque la sécrétion d’insuline.
Les sulfamides hypoglycémiants peuvent, de plus, inhiber la sécrétion de glucagon et sensibiliser les tissus cibles à l’action de l’insuline.
Les sulfamides hypoglycémiants s’administrent par voie orale, se fixent aux protéines plasmatiques, ont des demi-vies plasmatiques allant de cinq à dix heures, à l’exception du chlorpropamide et de la carbutamide où elle dépasse trente heures. Ils sont métabolisés au niveau hépatique.
Les sulfamides hypoglycémiants utilisés en thérapeutique peuvent être classés en fonction de leur durée d’action et de la quantité de produit actif efficace. Les nouveaux hypoglycémiants, glipizide, glibenclamide, gliclazide, glibornuride et glimépiride, sont actifs à doses beaucoup plus faibles que les anciens, tolbutamide, chlorpropamide qui ne sont plus commercialisés et carbutamide qui l’est encore.
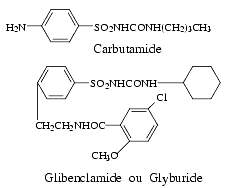
Utilisation
L’indication des sulfamides hypoglycémiants est le diabète de type II – c’est-à-dire ayant un pancréas capable de répondre à une stimulation par une augmentation de la sécrétion d’insuline – et non acido-cétosique et non équilibré par un régime approprié. Un bénéfice clinique résultant d’un traitement par sulfamides hypoglycémiants a été montré sur certaines manifestations, notamment les troubles rénaux et oculaires. Chez les diabétiques non insulinodépendants qui ont déjà une hyperinsulinémie, une sensibilisation des tissus à l’action de l’insuline par la metformine, par exemple, semble préférable à une stimulation de la sécrétion d’insuline.
|
Dénomination Commerciale |
||
Les sulfamides hypoglycémiants peuvent avoir des effets indésirables :
- Hypoglycémie, surtout en cas de surdosage par posologie trop élevée ou par inhibition de leur catabolisme ou par suppression d’un repas à la suite d’un exercice physique inhabituel
- Troubles digestifs (nausées, vomissements, cholestase)
- Troubles sanguins (anémie hémolytique)
- Réactions de type disulfirame ou Antabuse, surtout avec le chlorpropamide qui n’est plus commercialisé
- Hyponatrémies, par potentialisation de l’effet de l’hormone antidiurétique.
Glinides
D’autres substances n’ayant pas de groupe sulfamide comme la nateglinide et le repaglinide augmentent la sécrétion d’insuline par le même mécanisme d’action que les sulfamides. Leur effet hypoglycémiant est plus rapide et de plus courte durée que celui des sulfamides hypoglycémiants.
Le répaglinide a été le premier à être commercialisé en France. Pris avant les repas il évite l’hyperglycémie post-prandiale mais ses avantages et inconvénients à long terme par rapport aux sulfamides hypoglycémiants restent à préciser.
Les incrétinomimétiques sont des substances qui augmentent la libération d’insuline. Ce sont l’exénatique (BYETTA*), analogue GLP-1, les gliptines – sitagliptine (JANUVIA*). Voir Hormones d’origine digestive.
Potentialisateurs des effets de l’insuline
Les médicaments qui potentialisent les effets de l’insuline sont la metformine et les dérivés thiazolidinedione.
Metformine
La metformine est un biguanide. Elle diminue l’hyperglycémie sans risque d’hypoglycémie car elle n’abaisse pas la glycémie du sujet sain.
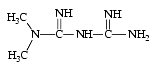
Son mécanisme d’action est complexe. Elle agit en présence d’insuline :
- en favorisant l’utilisation du glucose par les tissus, notamment par les muscles squelettiques
- en inhibant la néoglycogenèse hépatique, c’est-à-dire la formation du glycogène à partir des acides aminés et des lipides.
Les essais cliniques ont montré que la metformine réduisait chez les diabétiques la glycémie à jeun, l’hémoglobine glycosylée, la cholestérolémie et les triglycérides.
Contrairement aux sulfamides hypoglycémiants, la metformine ne stimule pas la sécrétion d’insuline. Elle peut donc être considérée comme un potentialisateur de l’effet de l’insuline.
La metformine n’est pas métabolisée par biotransformations. Elle est présente dans le plasma sous forme libre, non liée aux protéines. Sa demi-vie plasmatique est de l’ordre de deux à quatre heures. Elle s’élimine par le rein et, en cas d’insuffisance rénale, risque de s’accumuler. L’insuffisance rénale est donc une contre-indication à sa prescription.
Elle est indiquée dans le traitement du diabète de type II non équilibré par un régime approprié, particulièrement chez les sujets en surpoids. Elle est parfois utilisée comme adjuvant de l’insulinothérapie dans le traitement du diabète insulinodépendant. La metformine pourrait retarder la mortalité des diabétiques, surtout les obèses.
L’effet indésirable le plus grave de la metformine est l’acidose lactique, qui peut être mortelle, dont les signes prémonitoires sont des crampes, des troubles digestifs, des douleurs abdominales intenses, de l’asthénie. Ces signes doivent conduire à l’arrêt du traitement et à l’hospitalisation. Cette acidose lactique se voit surtout chez l’insuffisant rénal ou l’insuffisant hépatique. Le diagnostic est confirmé par le dosage de l’acide lactique, le prélèvement étant à faire sans garrot.
Elle peut avoir d’autres effets indésirables : troubles digestifs divers, nausées, vomissements, diarrhées, surtout en début de traitement.
La metformine doit être interrompue en cas d’examen radiologique par les agents de contraste iodés parce qu’ils sont hyperosmolaires et créent une déshydratation cellulaire, susceptible de favoriser l’acidose lactique.
|
GLUCOPHAGE* Cp à 500, 850 et 1000 mg |
Dérivés de structure thiazolidinedione
Le premier médicament du groupe des thiazolidinediones a été la troglitazone, dont l’activité se rapproche de celle de la metformine : elle ne stimule pas la sécrétion d’insuline mais potentialise son action, diminue l’hyperglycémie et la concentration sanguine d’hémoglobine glycosylée. Il s’agit d’un médicament intéressant mais dont la commercialisation a été arrêtée car il a été à l’origine d’hépatites graves.
Les nouveaux analogues de la troglitazone, pioglitazone et rosiglitazone, sont mieux tolérés. Leur intérêt par rapport à la metformine reste à préciser.
Les glitazones sont des agonistes des récepteurs PPAR gamma (Peroxisome Proliferator Activated Receptor gamma) qui, activés, forment des hétérodimères avec des récepteurs des rétinoïdes et modulent la trancription de gènes en ARN messager puis protéines (enzymes, transporteurs comme Glut-4) impliquées dans le métabolisme du glucose et des acides gras notamment. Elles stimulent la libération d’adiponectine par les adipocytes.
Formule chimique de la rosiglitazone avec l’explication du terme thiazolidinedione :

Au cours des études cliniques la rosiglitazone et la pioglitazone ont amélioré certains paramètres biologiques : diminution de l’hyperglycémie, de l’hémoglobine glycosylée, des acides gras libres plasmatiques. Mais elles n’ont pas réduit les complications du diabète ni la mortalité globale. Par ailleurs leurs effets indésirables: rétention hydrique et insuffisance cardiaque, troubles hépatiques (surveiller ALAT), prise de poids, anémie, notamment, ont conduit à les retirer du commerce. La rosiglitazone (AVANDIA*) a été retirée du commerce en septembre 2010 dans divers pays. La pioglitazone (Actos*) a été retirée du commerce en France en 2011. Pour plus de détails, voir ici et là.
Inhibiteurs de l’absorption digestive des glucides
Les a-glucosidases intestinales, situées au niveau de la bordure en brosse des entérocytes, libèrent le glucose par hydrolyse des résidus d’amidon, des oligosaccharides et des disaccharides.. Cette hydrolyse est nécessaire à l’absorption digestive des glucides car seuls les monosaccharides comme le glucose et le fructose sont absorbés.
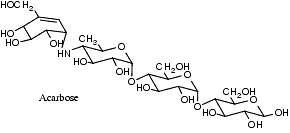
Si on inhibe les a-glucosidases par des produits comme l’acarbose, le miglitol ou l’émiglitate, on réduit et on retarde l’absorption digestive du glucose et, par là-même, l’hyperglycémie post-prandiale.
L’acarbose est un pseudotétrasaccharide qui se fixe avec une très grande affinité aux a-glucosidases et les inhibe. Son effet est réversible car il est hydrolysé et inactivé par les amylases et des enzymes bactériennes. L’acarbose est peu absorbé par le tube digestif.
Le miglitol comme l’acarbose agit au niveau de l’intestin mais il est absorbé par le tube digestif avec une biodisponibilité pouvant atteindre 90% lorsque la dose administrée est faible. La partie absorbée se distribue principalement dans l’espace extracellulaire et s’élimine par le rein.
On peut prescrire un inhibiteur des a-glucosidases dans le traitement du diabète non insulinodépendant, seul ou en association à d’autres antidiabétiques.
Le principal inconvénient de l’acarbose et du miglitol est de donner des troubles digestifs : flatulences, diarrhée, douleurs abdominales, surtout en début de traitement. L’acarbose a pu être exceptionnellement à l’origine d’états subocclusifs et d’hépatites.
Par ailleurs une alimentation enrichie en fibres végétales améliore le contrôle de la glycémie, diminue l’hyperinsulinémie et les lipides plasmatiques des diabétiques de type II.
Inhibiteurs de l’aldose réductase
Lorsque l’hyperglycémie provoque une augmentation des concentrations intracellulaires de glucose dans les tissus où sa pénétration est indépendante de l’insuline, l’excès de glucose non transformé en glucose-6-phosphate du fait de l’insuffisance de l’activité de la glucokinase, est réduit par l’aldose réductase en sorbitol et ensuite par la sorbitol déshydrogénase en fructose.
L’excès de sorbitol et de fructose intracellulaires altère la cellule, peut-être par un effet osmotique entraînant un appel d’eau et un gonflement cellulaire, avec rupture de la membrane plasmique. La déplétion de la cellule en taurine et en myoinositol qui intervient, notamment dans la synthèse de PIP2, participerait aussi à son altération.
L’inhibition de l’aldose réductase par des substances en cours d’étude comme le tolrestat, l’imirestat, le ponalrestat, diminuerait certaines conséquences néfastes de l’hyperglycémie.
Une supplémentation par la vitamine C, même à doses faibles, de l’ordre de 100 mg/jour, inhiberait l’aldose réductase et s’opposerait à l’accumulation de sorbitol dans les érythrocytes mais ce résultat ne semble pas avoir été confirmé.
