Certains antiviraux agissent en perturbant la replication de l’ADN, DNA, viral ou de l’ARN, RNA, viral par inhibition soit de la DNA polymérase, soit de la RNA polymérase ou de la transcriptase inverse ou de certaines protéines impliquées dans la replication virale. Beaucoup d’entre eux sont des analogues des bases puriques ou pyrimidiques et appelés inhibiteurs nucléosidiques, et agissent après avoir été phosphorylés mais d’autres inhibiteurs sont actifs d’emblée sans phosphorylation préalable, ce sont des inhibiteurs non nucléosidiques.
Inhibiteurs de la transcriptase inverse du VIH
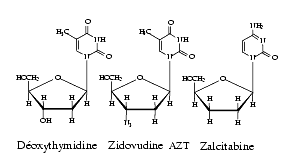
Les inhibiteurs nucléosidiques de la transcriptase inverse, appelée aussi réverse transcriptase, actifs contre le virus de l’immunodéficience humaine ou VIH, virus à RNA sont des analogues des bases pyrimidiques et puriques. Ce sont la zidovudine, la zalcitabine, la stavudine, la lamivudine, la didanosine, l’emtricitabine, l’abacavir, et le ténofovir. Ils agissent après métabolisation en dérivés triphosphates.
Zidovudine
La zidovudine, ou 3′-azido-3′-déoxythymidine ou AZT, molécule connue depuis 1964, est un analogue structural de la déoxythymidine : le groupe OH en 3′ du déoxyribose ayant été remplacé par le groupe azido N3. La zidovudine, en raison de l’absence de groupe OH en 3′, inhibe la transcriptase inverse, empêche l’élongation de la chaîne de DNA, réplique du RNA viral.
La zidovudine n’est active qu’après sa phosphorylation en AZT triphosphate qui s’effectue dans les cellules saines et infectées. L’AZT triphosphate est à la fois substrat et inhibiteur de la transcriptase inverse, enzyme responsable la synthèse de DNA à partir de RNA des virus de l’immuno-déficience humaine de type 1 et 2.
La zidovudine a plus d’affinité pour la transcriptase inverse virale que pour la DNA polymérase humaine. La transcriptase inverse catalyse à partir d’un simple brin de RNA viral la formation d’un double brin de DNA, lequel, grâce à la machinerie de la cellule hôte, sera à l’origine de la synthèse de RNA viral.
Les témoins de l’efficacité d’un médicament anti-SIDA sont la remontée du nombre de lymphocytes CD4, marqueur de l’état immunitaire, qui chute au cours de la maladie, et la diminution de la charge virale, concentration de RNA viral circulant, marqueur de l’intensité de la replication du virus dans l’organisme.
L’action antivirale de la zidovudine est antagonisée par l’apport de thymidine ou de ribavirine.
Sur le plan pharmacocinétique, l’AZT est absorbée à environ 70% par le tube digestif, sa demi-vie plasmatique est courte, de l’ordre d’une heure, ce qui explique la nécessité de prises répétées toutes les quatre heures. Le probénécide prolonge la demi-vie de l’AZT en réduisant son élimination rénale.
Les indications classiques de l’AZT sont le traitement du syndrome d’immuno-déficience acquise ou SIDA, et le traitement préventif des malades à sérologie HIV positive pour tenter de retarder l’évolution vers la maladie. L’AZT est également utilisée pendant la grossesse pour tenter de réduire la transmission du virus de la mère à l’enfant. L’utilisation de zidovudine pendant le premier trimestre de la grossesse ne semblait pas augmenter le risque de malformations de l’enfant. Toutefois il est apparu que les enfants nés de femmes enceintes traitées par AZT pouvaient avoir des anomalies mitochondriales avec troubles neurologiques ou cardiaques et il a été recommandé de ne commencer le traitement par AZT qu’après le troisième trimestre de la grossesse.
Depuis que l’on dispose d’autres médicaments inhibiteurs de la transcriptase inverse et de la protéase à activité post-translationnelle, le principe du traitement est l’association de trois médicaments et est appelée trithérapie.
L’AZT a plusieurs effets indésirables :
- hématologiques : anémie, neutropénie
- digestifs : nausées, vomissements, douleurs abdominales
- neurologiques : céphalées, insomnie, myalgies, asthénie.
La prise simultanée d’AZT et de paracétamol augmente l’incidence des neutropénies.
Les inhibiteurs nucléosidiques de la transcriptase inverse du VIH, outre la zidovudine, sont abacavir, didanosine, emtricitabine, lamivudine, stavudine et ténofovir.
| RETROVIR* | |
|
Abacavir |
ZIAGEN* |
|
Didanosine |
VIDEX* |
|
Emtricitabine |
EMTRIVA* |
|
Lamivudine |
EPIVIR* |
|
Stavudine |
ZERIT* |
|
Ténofovir |
VIREAD* |
La lamivudine ou déoxythiacytidine ou 3TC est un analogue nucléosidique où un atome de carbone du noyau ribose a été remplacé par un atome de soufre. C’est un inhibiteur de la transcriptase inverse utilisé dans le traitement des infections à VIH et dans le traitement de l’hépatite B chronique évolutive. Ses principaux effets indésirables sont hématologiques (anémie, neutropénie) et hépatiques (élévation des transaminases).La stavudine ou didéhydro-déoxythymidine ou d4T agit par ses métabolites phosphorylés et inhibe la transcriptase inverse. Son effet indésirable le plus marquant est une neuropathie périphérique dose-dépendante. Elle peut aussi donner des pancréatites. Son association à la zidovudine (RETROVIR*) n’est pas conseillée car la zidovudine pourrait inhiber la phosphorylation intracellulaire de la stavudine et la rendre moins efficace.
La didanosine (2′-3′-didéoxyinosine) ou ddI, analogue des bases puriques, inhibe, outre la transcriptase inverse, l’élongation de la chaîne des acides nucléiques viraux. Parmi les effets indésirables de la didanosine, il faut citer
les neuropathies périphériques et les pancréatites.
L’abacavir, analogue de la guanosine, est un inhibiteur nucléosidique de la transcriptase inverse. Actif contre le VIH 1 et le VIH 2, il est utilisé dans le traitement du SIDA. Il peut être à l’origine de manifestations l’hypersensibilité,
notamment respiratoires, telles que dyspnée, bronchospasme, les plus graves pouvant mettre en jeu le pronostic vital. Ces réactions doivent conduire à l’arrêt de l’abacavir et à sa non-utilisation ultérieure.
Le ténofovir, analogue nucléosidique monophosphate, c’est un nucléotide, qui agit après transformation en dérivé diphosphate.
L’emtricitabine est un analogue nucléosidique de la cytosine, dont la structure est proche de celle de la lamivudine. Elle est active contre le VIH1 et le VIH2 et le virus de l’hépatite B.
Les inhibiteurs non nucléosidiques de la transcriptase inverse du VIH sont l’éfavirenz, l’étravirine, la névirapine et la rilpivirine.
|
Efavirenz |
SUSTIVA* |
|
Etravirine |
INTELENCE* |
|
Névirapine |
VIRAMMUNE* |
|
Rilpivérine |
EDURANT* |
La névirapine est un inhibiteur non nucléosidique et non compétitif de la transcriptase inverse du VIH-1 mais non du VIH-2. Elle inhibe également la DNA polymérase. Une résistance à son effet apparaît très rapidement lorsqu’elle est utilisée seule. Elle a a une bonne biodisponibilité (> à 90%) et une longue demi-vie, environ 30 h.
Ses principaux effets indésirables sont les éruptions cutanées parfois très graves, fièvre, hépatites.
L’éfavirenz est un inhibiteur non nucléosidique de la transcriptase inverse du VIH-1 mais non VIH-2, directement actif sans devoir être métabolisé en dérivé triphosphate. Sa biodisponibilité est peu modifiée par la prise concomitante d’aliments, à l’exception des lipides qui l’augmentent. Il est métabolisé en métabolites inactifs principalement par les cytochromes CYP3A et CYP2B6. Il est inducteur enzymatique mais les conséquences éventuelles de cet effet restent à évaluer. Il est utilisé dans le traitement de l’infection par le VIH-1, en association avec d’autres médicaments. L’efavirenz augmenterait la tendance suicidaire.
Associations de 2 ou 3 principes actifs: en pratique, la base du traitement de l’infection par le VIH comporte une trithérapie avec, par exemple deux inhibiteurs de la transcriptase inverse et un autre type d’inhibiteur. Il existe plusieurs spécialités associant 2 ou 3 principes actifs anti-VIH destinées à réduire le nombre de comprimés à prendre quotidiennement.
|
Zidovudine (300 mg) + |
COMBIVIR* Cp |
|
Abacavir + Lamivudine |
KIVEXA* Cp |
|
Abacavir + Lamivudine + |
TRIZIVIR* Cp |
|
Ténofovir + Emtricitabine |
TRUVADA* Cp |
|
Ténofovir + Emtricitabine + |
ATRIPLA* Cp |
|
Ténofovir + Emtricitabine + |
EVIPLERA* Cp |
Remarque
Les inhibiteurs de la transcripase inverse ont été les premiers antiviraux efficaces contre le VIH et demeurent les plus utilisés. D’autres médicaments anti-VIH sont venus les compléter :
- les inhibiteurs de l’adhérence du virus à la cellule-hôte type maraviroc,
- les inhibiteurs de la fusion des membranes type enfuvirtide,
- les inhibiteurs de la protéase type ritonavir,
- les inhibiteurs de l’intégrase type raltégravir.
Inhibiteurs de l’ARN polymérase du virus de l’hépatite C, VHC
Le sofosbuvir est un inhibiteur de l’ARN polymérase ARN dépendante NS5B, replicase assurant la copie de l’ARN du virus de l’hépatite C. Le sofosbuvir est une prodrogue qui dans l’organisme est convertie en un analogue de l’uridine triphosphate qui incorporé dans l’ARN viral perturbe son fonctionnement. Il est indiqué dans le traitement de l’hépatite C chronique. Il apporte un progrès remarquable mais est très coûteux.
Les autres produits actifs contre le virus de l’hépatite C sont le daclatasvir, le dasabuvir, le siméprevir, l’ombitasvir et le ledipasvir
|
Sofosbuvir |
SOVALDI* Cp 400 mg |
|
Siméprevir |
OLYSIO* Cp |
|
Dasabuvir |
EXVIERA* Cp |
|
Daclatasvir |
DAKLINZA* Cp |
|
Ledispasvir + Sofosbuvir |
HARVONI* Cp |
|
Ombitasvir + Paritaprevir + Ritonavir |
VIEKIRAX* Cp |
Inhibiteurs de l’ADN-polymérase du virus de l’hépatite B, VHB
Le virus de l’hépatite B, VHB, est un virus à ADN. Sa polymérase fonctionne comme ADN dépendante à partir de la matrice d’ADN (complétant l’ADN viral partiellement bicaténaire en ADN bicaténaire) et comme ARN dépendante à partir de la matrice ARN, c’est-à-dire une transcriptase inverse, ce qui explique que certains inhibiteurs de la transcriptase inverse du VIH puissent inhiber la replication du VHB.
Les inhibiteurs de la polymérase du VHB sont l’adéfovir disoproxil, la lamivudine, l’entécavir, ténofovir et la telbivudine.
Le ténofovir, analogue nucléosidique monophosphate, c’est un nucléotide, qui agit après transformation en dérivé diphosphate.
L’entecavir est un analogue structural de la guanosine nucléoside. Initialement développé comme antiherpétique, il s’est révélé peu actif dans cette indication mais très actif contre le virus de l’hépatite B, VHB.
|
Adéfovir |
HEPSERA* Cp |
|
Lamivudine |
ZEFFIX* Cp |
|
Entécavir |
BARACLUDE* Cp |
|
Ténofovir |
VIREAD* Cp |
|
Telbivudine |
SEBIVO* Cp |
Inhibiteurs de l’ADN-polymérase du virus herpétique
Les analogues des bases puriques et pyrimidiques actifs contre les virus à DNA type herpès sont l’aciclovir, le valaciclovir, le famciclovir, l’idoxuridine, la trifluridine, la vidarabine, le ganciclovir et le cidofovir Ils sont utilisés dans le traitement des infections à virus de la famille Herpes viridae. Il s’agit de l’herpes simplex, de l’herpes zoster (zona) et des infections à cytomégalovirus.
L’aciclovir, qui s’écrit parfois acyclovir, est un analogue de la guanine substitué en position 9 par remplacement d’un atome d’hydrogène par une chaîne à fonction alcool. Pour être actif, l’aciclovir doit être phosphorylé en aciclovir triphosphate qui entre en compétition avec la déoxyguanosine triphosphate.
Les biotransformations subies par l’aciclovir dépendent de la présence de thymidine kinase virale à l’intérieur de la cellule : en absence d’infection, l’aciclovir n’est pas transformé en aciclovir monophosphate et les étapes ultérieures ne sont donc pas possibles.
En présence de virus à l’intérieur de la cellule, l’aciclovir est transformé en aciclovir monophosphate puis, sous l’influence des enzymes cellulaires, en aciclovir diphosphate et triphosphate. L’aciclovir triphosphate inhibe la DNA polymérase et empêche la multiplication virale.
La spécificité de l’action de l’aciclovir vient donc du fait qu’il est phosphorylé par la thymidine kinase du virus de l’herpès et non par la thymidine kinase de la cellule de l’hôte, si bien que l’on trouve beaucoup d’aciclovir sous forme active, c’est-à-dire triphosphate, dans les cellules infectées et pas dans les cellules non infectées.
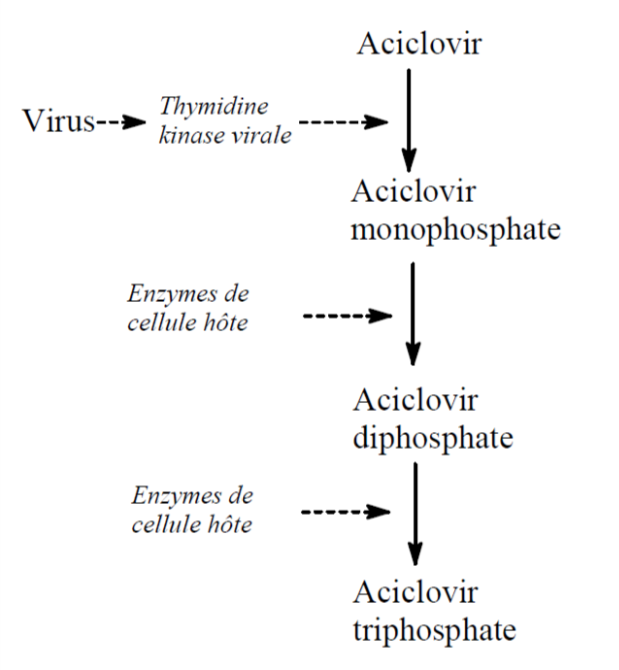
Sur le plan pharmacocinétique, la biodisponibilité de l’aciclovir par voie orale est faible, 15 à 30%; sa demi-vie plasmatique environ deux heures; son élimination rénale.
Ses indications sont essentiellement le traitement des infections à virus varicelle-zona. Les posologies utilisées dans le traitement préventif (voie buccale) sont beaucoup plus faibles que celles qui sont utilisées en traitement curatif (voie parentérale en général). L’aciclovir est utilisé sous forme de crème pour traiter l’herpès labial. L’aciclovir n’a pas montré d’efficacité réelle dans le traitement de la mononucléose infectieuse due au virus Epstein-Barr.
|
Aciclovir |
ZOVIRAX* Cp 200 et 800 mg, Inj, Pommade, crème |
Le valaciclovir, constitué d’une molécule d’aciclovir estérifiée par la L-valine, a une biodisponibilité par voie orale nettement plus importante que celle de l’aciclovir. Après absorption, il est hydrolysé en aciclovir. Il est utilisé dans la prévention des douleurs associées au zona et la prévention des complications oculaires du zona ophtalmiqus. Pour être efficace le traitement doit être débuté dans les 72 heures qui suivent l’apparition de l’éruption.
|
Valaciclovir |
ZELITREX* Cp |
Le famciclovir s’administre par voie orale et est le précurseur du penciclovir, dérivé purique de structure proche de celle de l’aciclovir. Il s’utilise dans le traitement des infections à virus Herpes simplex et à virus du zona dont la prévention des complications du zona ophtalmique. Il agit après transformation en penciclovir triphosphate.
|
Famciclovir |
ORAVIR* Cp |
La trifluridine, ou 5-trifluorométhyl-2′-déoxyuridine, ou trifluorothymidine, inhibe, après sa transformation en trifluridine triphosphate, le DNA viral dans lequel elle s’incorpore. Elle est utilisée sous forme de collyre dans le traitement des atteintes oculaires herpétiques.
|
Trifluridine |
VIROPHTA* Collyre |
Le ganciclovir a une structure très proche de celle de l’aciclovir et, comme lui, est actif après sa métabolisation en dérivé triphosphate, mais il a une activité antivirale plus importante sur les cytomégalovirus qui constituent un sous-groupe de virus de type herpès. Il est utilisé pour traiter les diverses infections à cytomégalovirus, en particulier la rétinite qui s’observe chez les malades atteints de SIDA. Le ganciclovir existe sous des formes destinées à être administrées par voie générale, buccale et intraveineuse et par voie locale, oculaire.
|
Ganciclovir |
CYMÉVAN* Inj |
La forme orale a une biodisponibilité d’environ 6%, mais elle est augmentée jusqu’à environ 20% par la prise concomitante d’aliments.
La toxicité hématologique du ganciclovir en administration par voie générale est importante et se traduit chez environ un tiers des malades traités par une neutropénie ou une thrombopénie. Il est tératogène. Utilisé par voie intraveineuse, il pourrait entraîner chez l’homme et chez la femme une stérilité temporaire ou même définitive.
Il existe une présentation ophtalmique destinée au traitement de la kératite herpétique superficielle et une présentation implant oculaire destinée au traitement local de la rétinite à cytomégalovirus des malades infectés par le VIH. La mise en place de cet implant nécessite une intervention chirurgicale.
|
Ganciclovir |
VIRGAN* Gel ophtalmique |
Le valganciclovir est métabolisé dans l’organisme en ganciclovir. Il a une biodisponibilité par voie orale d’environ 60 %, 10 fois plus élevée que celle du ganciclovir.
|
Valganciclovir |
ROVALCYTE* Cp, Sol buv |
Le cidofovir est un analogue de la cytidine, actif après phosphorylation contre les souches de cytomégalovirus humain résistant au ganciclovir. Il est prescrit dans le traitement de la rétinite à cytomégalovirus chez les immunodéficients.
|
Cidofovir |
VISTIDE* Inj (perfusion) |
