Auteurs :
Hélène Berthelot (Banques de Données sur le Médicament Consultant (BDMC))
Gérard Simon (Pharmacien Consultant)
Massoud Toussi (IMS Health, France)
Les erreurs médicamenteuses, leurs conséquences et les mesures permettant leur prévention font partie des préoccupations des décideurs de santé publique. En France, le nombre de décès qui sont les conséquences de ces erreurs atteindrait 10 000 par an (1). Aux Etats-Unis, on estime entre 44 000 et 98 000 le nombre de patients qui meurent annuellement du fait d’erreurs médicamenteuses évitables (2).
Selon une définition proposée dans le cadre d’une rencontre organisée par l’Agence Européenne de Médecine en février 2013 (3), l’erreur médicamenteuse est définie comme « une défaillance dans le processus de traitement, que ce soit par excès ou par défaut, qui mène à, ou a le potentiel de causer un préjudice pour le patient». Une étude importante réalisée aux Royaume-Uni, a récemment montré que les erreurs d’administration, de prescription et de dispensation constituent plus de 80% des erreurs médicamenteuses (4). En ce qui concerne les catégories d’erreur, la même étude montre que les plus courantes portent sur les omissions ou retards de traitement, le choix du médicament et de son dosage, la fréquence d’utilisation ou la dose. En ce qui concerne les médicaments les plus fréquemment affectés, les opioïdes sont en tête de liste, suivi par les antibiotiques, antivitamines K, héparines à bas poids moléculaire, insulines, benzodiazépines et anti-inflammatoires non stéroïdiens.
En France, selon le guichet des erreurs médicamenteuses, les erreurs de dosage représentent 7,3 % des signalements, les erreurs de posologie 27,7 %, les erreurs de médicaments 41,8 %, les erreurs de voie d’administration 5,5 % (5).
Les Logiciels d’Aide à la Prescription (LAPs) ont été introduits en partie pour réduire les erreurs médicamenteuses. De très nombreuses publications ont étudié l’influence de ces logiciels sur la réduction des erreurs de prescription. Cependant, si les auteurs ont observé des améliorations, ils ont parfois observé une non réduction signifiante des erreurs attribuables à ces logiciels, voire même une augmentation des erreurs liée à leur utilisation (6–8). La plupart des études réalisées fournissent des données statistiques afin de classer par ordre décroissant de prévalence les types d’erreurs de prescription les plus importantes observées. Les intitulés des types d’erreurs signalés et leur signification n’étant cependant pas homogène, nous n’avons pu en tirer une analyse statistique globale. Tout au plus peut on proposer une liste récapitulative par fréquence décroissante des erreurs :
- Erreurs de médicament
- Erreurs de posologies
- Erreurs de voie d’administration
- Erreurs d’unités d’administration
- Erreurs de durée de traitement
- Interactions médicamenteuses
- Prise en compte insuffisante des données physiopathologiques du patient
Il est remarquable de constater la majorité des erreurs de prescription décrites dans la littérature concernent l’hôpital et émanent de pharmaciens hospitaliers rompus à la fonction d’analyse pharmaceutique des prescriptions. La littérature est par contre beaucoup plus rare en médecine de ville. Plusieurs causes possibles à ce silence dont la principale est sans doute le peu de temps dont dispose le pharmacien d’officine pour analyser la prescription médicale et les processus moins rigoureux obligeant une revue systématique des prescriptions médicales par le pharmacien de ville.
Aussi, avec l’utilisation de plus en plus répandue des LAPs, les médecins prennent l’habitude d’exploiter ce que leur propose leur logiciel en matière d’information sur les médicaments et la lisibilité de leurs libellés, en particulier en matière de dosages, interactions médicamenteuses, posologies (durée de traitement, unités de prises et voies d’administration recommandées). Ceci nécessite une recherche proactive des risques de défaillances pouvant survenir dans le processus de prescription informatisée, afin de pouvoir les éviter par la mise en place des mesures de minimisation de risque.
Les conséquences des défaillances pour le patient, si elles ne sont pas détectées par le prescripteur, sont de deux ordres :
- Un défaut d’alertes du LAP durant la prescription pouvant conduire à une prescription potentiellement dangereuse.
- Un excès d’alertes ou des alertes de niveau de gravité exagéré pouvant conduire à la non prescription d’un médicament nécessaire cependant à la guérison du patient. Cette conséquence n’apparaît pas dans la littérature car n’étant pas détectée ni classée dans la rubrique « erreurs médicamenteuses ».
Afin d’améliorer la sécurité de la prescription médicamenteuse, la Haute Autorité de Santé (HAS) a élaboré deux procédures : l’Agrément des Banques de données sur le Médicament (BdMs) et la certification des LAPs et des Logiciels d’Aide à la Dispensation (LADs)
L’agrément des BdMs est accordé après l’engagement de leurs éditeurs au respect d’une Charte de Qualité et réponse à un questionnaire dont les réponses sont évaluées par un groupe d’experts. Cette mesure est une étape significative en matière d’amélioration de la sécurité mais n’implique ni examen réel du contenu des BdMs ni surveillance de la conformité des réponses au questionnaire. Le contrôle de la qualité effective est laissé à la discrétion des utilisateurs finaux pouvant signaler leurs observations à la HAS.
La certification des LAPs qui inclut l’obligation pour ceux-ci d’utiliser une BdM agréée a pour objet de vérifier la présence effective de certaines fonctions d’alerte et de prévention de la iatrogénie.
Cependant, si ces deux procédures se sont avérées nécessaires, elles ne sont pas suffisantes pour garantir la sécurité car aucun contrôle de la qualité du contenu des informations et de leur conformité aux sources référentielles n’est réalisé, que ce soit au niveau des BdMs ou à celui des LAPs et / ou des LADs.
Une publication de 1998, analysant 23.000 prescriptions et les confrontant aux données issues d’une BdM a décrit des indicateurs quantitatifs permettant de rendre compte de la qualité des prescriptions en matière de prévention de la iatrogénie (14).
Une étude récente a tenté, à l’aide de prescriptions-tests, d’évaluer la conformité du contenu des BdMs aux résultats théoriques attendus. Elle a conclu à un taux de conformité situé entre 20 et 30% sans différence significative entre les BdMs étudiées (Claude Bernard, Thériaque, Thesorimed et Vidal) (10).
Nous avons utilisé la méthode AMDEC pour identifier pro-activement les modes de défaillance de la chaine de prescription informatisée.
L’objectif de notre recherche était d’identifier les modes de défaillances d’un processus de prescription informatisée en mettant l’accent sur les outils informatiques tels que les BdMs et LAPs, hiérarchiser les modes de défaillances en fonction de leur criticité et proposer des recommandations afin de réduire les risques liés à l’utilisation de ces logiciels.
Méthode
L’Analyse des modes de défaillance (formes observables d’un dysfonctionnement), de leurs effets et de leur criticité (AMDEC) est une méthode utilisée en industrie pour évaluer les processus complexes selon une approche standardisée d’identification des éléments à risque. Il est basé sur le principe que le risque lié à une défaillance n’est pas seulement lié à sa fréquence, mais aussi à son impact et à la difficulté de sa détection. Cette approche permet également de hiérarchiser numériquement les risques identifiés selon le produit de ces trois facteurs (fréquence, impact et probabilité de non détection).
Bien que cette méthode soit utilisée dans le domaine militaire depuis la deuxième guerre mondiale, son utilisation dans le domaine de la santé est plus récente (11). LAGO et al l’ont utilisé en 2012 pour l’analyse et la réduction des risques d’erreurs de prescription dans les services de pédiatrie en Italie sans mettre l’accent sur les processus informatisés. Ils ont montré que son utilisation a permis la détection de 37 modes de défaillances à haute priorité. L’introduction proactive des solutions de prévention a permis de réduire de 60% les modes de défaillances à haut risque au deuxième passage de la méthode (12).
Mise place d’une équipe pluridisciplinaire
L’équipe d’experts comprend deux pharmaciens co-créateurs d’une des quatre BdMs (Base Claude-Bernard – BCB. Les autres BdMs sont Thériaque, Thésorimed, et Vidal) agrées et désormais experts indépendants et d’un médecin informaticien, certains ayant participé aux commissions d’agrément des BdMs et de certifications des LAPs et LADs de la HAS.
Les responsabilités de l’équipe étaient de :
- Réaliser une cartographie des différentes étapes de transformation des données dans un processus de prescription et de délivrance informatisée ;
- Identifier les modes de défaillance (sources potentielles d’erreur) dans un tel processus et en déterminer les causes ;
- Définir le niveau de criticité de l’impact de chaque mode de défaillance identifié ;
- Proposer des mesures correctives ou préventives pour chaque mode de défaillance et recalculer son niveau de criticité prenant en compte ces mesures.
Cartographie des étapes du processus de prescription ou de délivrance informatisée
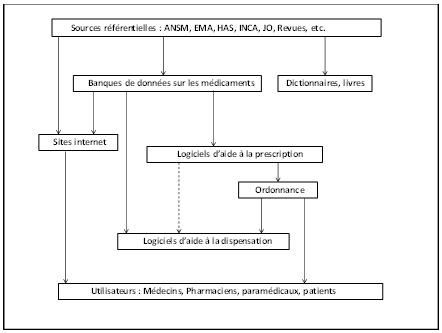
Les étapes successives de transformation de l’information sur le médicament depuis les sources primaires jusqu’à la dispensation ont été décrites. La cartographie schématique du processus a permis d’envisager les différents modes de défaillance pouvant survenir à chacune des étapes.
Identification des modes de défaillance
Les plus fréquents des modes de défaillance pouvant survenir dans le processus de prescription ou de dispensation informatisées ont été regroupés dans une des catégories suivantes :
- Choix du médicament,
- Interactions médicamenteuses,
- Posologies,
- Grossesse et Allaitement,
- Contre-indications par rapport au profil physiopathologique du patient,
- Prescription en Dénomination Commune (DC).
Pour chaque mode de défaillance, une ou plusieurs causes possibles ont été précisées, plusieurs erreurs effectivement constatées, les acteurs probables identifiés et des recommandations proposées.
Pondération des modes de défaillance
Le niveau de criticité des modes de défaillance a été calculé à l’aide de trois indicateurs :
- La probabilité de l’occurrence (P), définie sur trois niveaux :
- Fréquent : la défaillance pourrait concerner un grand nombre de prescriptions (supérieure à 10%).
- Occasionnel : la défaillance pourrait concerner un nombre plus faible mais non négligeable des prescriptions (entre 0,01% et 10%)
- Rare : la défaillance pourrait concerner un nombre anecdotique de prescriptions (inférieure à 0,01%)
- La sévérité de l’impact (S), définie sur trois niveaux :
- Grave : la défaillance est susceptible d’induire un effet indésirable plus ou moins invalidant pouvant nécessiter une hospitalisation voire provoquer un décès.
- Moyen : la défaillance ne permet pas s’assurer l’efficacité optimale du traitement.
- Faible : la défaillance est sans conséquence sur la santé du patient.
- La facilité de détection (D), définie sur trois niveaux :
- Impossible : en cas de défaillance, les étapes suivantes du processus ne pourraient pas permettre de détecter l’anomalie.
- Difficile : en cas de défaillance, les étapes suivantes du processus pourraient permettre de détecter l’anomalie si vigilance particulière.
- Evidente : en cas de défaillance, les étapes suivantes du processus permettraient de détecter l’anomalie.
Pour chaque indicateur, le score de 1, 3 ou 9 a été attribué selon le niveau (Tableau 1).
| Score |
Probabilité (P) |
Sévérité (S) |
Détection (D) |
|
1 |
Rare |
Faible |
Evidente |
|
3 |
Occasionnel |
Moyen |
Difficile |
|
9 |
Fréquent |
Grave |
Impossible |
Tableau 1 : Le score attribué à chaque niveau de probabilité, sévérité ou détection
Une fois les scores P, S et D attribués à chaque mode de défaillance, la criticité du mode de défaillance (Risk Priority Number, RPN) a été calculée en utilisant la formule suivante : RPN = P X S X D
Proposition des mesures correctives ou préventives
Pour chaque mode de défaillance identifié, une ou plusieurs mesures correctives ou préventives ont été proposées, puis la criticité a été recalculée en prenant en compte ces mesures. Cet exercice a permis d’identifier les solutions les plus intéressantes, permettant d’obtenir le gain le plus important en termes de RPN. Le Tableau 2 montre le modèle utilisé par le groupe pour la proposition des modes de défaillance, le calcul de leur criticité avant et après les solutions.
|
Risque |
Cause |
Exemples |
Acteurs |
P |
S |
D |
RPN |
Solution (corrective ou préventive) |
P |
S |
D |
RPN |
gain |
Résultats
Cartographie des étapes du processus de prescription ou délivrance informatisé
Première étape : les sources primaires référentielles
Le fondement opposable de l’information sur le médicament est constitué de sources primaires et référentielles disponibles sur les sites des instances réglementaires en format textuel où les connaissances sont exprimées par des mots, des phrases, du vocabulaire commun que les professionnels à qui elles sont destinées, peuvent traiter sans difficulté. L’intellect humain peut facilement comprendre les informations implicites, non formalisées, les interpréter, les hiérarchiser et les nuancer (13).
En France, les sources concernées sont les suivantes (liste non exhaustive) :
- L’ANSM pour :
- Le Résumé des Caractéristiques Produit (RCP) et la Notice d’Information pour le patient (NIP) ;
- Le fichier des compositions, présentations et spécialités ;
- Le thésaurus des Interactions Médicamenteuses (IAM) ;
- Le répertoire des génériques ;
- Le livret des substances actives en lien avec le déficit en glucose-6-phosphate déshydrogénase (G6PD) ;
- Le Guideline des excipients à effet notoire (EEN) ;
- Les importations et distributions parallèles ;
- Les Autorisations Temporaires d’Utilisation (ATU) de Cohorte et Nominatives.
- La HAS pour les avis de la Commission de la Transparence (CT) et les avis de synthèse.
- L’Agence Européenne pour les Médicaments (EMA) pour les RCPs, Etiquetage et Notices des spécialités avec AMM européenne.
- L’Institut National Cancer (INCA) pour les protocoles thérapeutiques.
- Le Journal Officiel pour les données économiques sur le médicament, les fiches thérapeutiques, les produits dopants, les pictogrammes conduite.
A ces sources textuelles, il convient d’ajouter des nomenclatures officielles :
- La « Classification Internationale des Maladies » (CIM) dans sa dixième édition (CIM.10) ;
- Les « Standard Terms », nomenclature des formes galéniques et des voies d’administration de la pharmacopée européenne ;
- La nomenclature des principes actifs (Non Proprietary Names) de l’OMS ;
- La classification Anatomique, Thérapeutique et Chimique (ATC).
A coté de ces sources primaires, il convient de citer une autre source d’informations émanant des laboratoires pharmaceutiques à savoir les mentions légales. Ces mentions légales ne constituent pas une source opposable au sens légal du terme car ne recevant pas l’imprimatur de l’ANSM.
D’autres sources primaires sont constituées par des organismes, sans lien avec l’industrie, mais ne constituant pas pour autant, quelle que soit leur qualité, des sources opposables telles que Le Centre de Référence sur les Agents Tératogènes (CRAT) (grossesse et allaitement).
Enfin, plus largement, des sources scientifiques classiques telles que les articles, les synthèses de la littérature, les guides de bonnes pratiques cliniques. Ces informations, notamment celles opposables, sont d’accès libre et permettent aux professionnels de santé une consultation en ligne détaillée lors de la prescription ou de la dispensation. Les pharmaciens hospitaliers exerçant une fonction d’analyse pharmaceutique des prescriptions se réfèrent préférentiellement à ces sources ainsi qu’à la littérature scientifique mais, en médecine ambulatoire et en pharmacie de ville, le temps nécessaire à cette analyse faisant défaut explique l’utilisation croissante des outils de détection inclus dans les LAPs et LADs.
Dans ces cas, les failles de l’information référentielle risquent d’être présentes dans les BdMs puis, sauf correction improbable par les éditeurs de logiciels dans les LAPs et LADs.
La probabilité de détection de l’erreur est alors très faible car si les BdMs et logiciels la reproduisent, l’absence de contradiction entre les différents moyens informatiques utilisables par les professionnels consolide l’erreur.
Seconde étape : la génération des Banques de données (sources secondaires)
La transformation de l’information est mise en œuvre par les BdMs. A cette étape, par un travail d’auteur, l’information textuelle originelle (comme les RCPs des médicaments) fait l’objet d’une simplification et d’une structuration en connaissances formelles, exprimées sous forme d’objets logiques, structurés, où chaque élément doit être explicite et codifié afin de la rendre utilisable par des outils informatiques.
La difficulté de cette structuration de l’information tient à la nécessité de résumer des chapitres tels que ceux des indications ou des contre-indications où sont mentionnées notamment des pathologies mais parfois aussi des situations cliniques complexes. Les textes sont alors résumés et représentés par un ou plusieurs mots-clés, interfacés ensuite avec des mots et codes de nomenclatures telles que la CIM.10. Cette étape est critique car cette transformation radicale de l’information implique un travail de synthèse mettant en jeu des compétences pointues dans divers domaines médico-pharmaceutiques et informatiques.
Cette transformation de l’information primaire en données structurées doit en outre prendre en compte l’interopérabilité nécessaire avec les données structurées du dossier patient informatisé (DPI) lorsque celui-ci comporte certains éléments tels que l’âge, le sexe, le poids, la taille, le statut de grossesse, l’allaitement, les allergies, les pathologies, les antécédents personnels ou familiaux de pathologie, les données de biologie telles que la clairance de la créatinine pour l’insuffisance rénale.
Les risques de dégradation, de simplification, d’erreurs d’interprétation lors de cette étape sont des risques majeurs actuellement mal cernés. .
Troisième étape : les logiciels (sources tertiaires)
La troisième étape du transfert de l’information est celle des LAPs et des LADs exploitant la (ou les) BdM(s) afin de générer, durant le travail du professionnel de santé, des alertes de prévention de la iatrogénie en utilisant les objets informatiques fournis par les BdMs et en croisant ces informations avec les données issues du dossier patient.
Cette série de transferts et de transformation de l’information sur le médicament nécessite des interventions humaines nombreuses portant tout à la fois sur l’exploitation du contenu pharmaco-clinique fourni par les BdMs que sur le développement des outils informatiques, de lecture et de consultation.
L’intégration de la BdM dans un logiciel peut s’opérer de diverses manières :
- Par le moyen d’une bibliothèque de fonctions (Application Programming Interface, API) conçue par l’éditeur de la BdM et intégrée directement par l’éditeur de LAP ;
- Par le moyen d’une bibliothèque de fonctions en open source permettant à l’éditeur de logiciel d’adapter chacune d’elles à ses objectifs ;
- Par l’utilisation directe des tables informatiques fournies par la BdM.
Il peut d’ailleurs exister un panachage de ces méthodes.
L’intégration de la BdM dans le LAP ou le LAD implique un processus informatique nécessitant des compétences élargies et dont la complexité peut générer des erreurs. En effet, si l’éditeur du logiciel utilise des tables, il doit lui-même écrire les programmes de génération d’alertes, ce qui suppose des connaissances pharmacologiques.
Dans le meilleur des cas, lorsque la BdM fournit une bibliothèque de fonctions élaborée, l’éditeur doit assurer l’interopérabilité entre les données structurées du patient et les API, avec le risque de non-reconnaissance de codes ou d’expressions.
Les LAPs et les LADs intégrant sans les modifier les données structurées fournies par les BdMs, les erreurs éventuelles de ces dernières seront encore présentes dans les logiciels utilisés par les professionnels de santé.
Quatrième étape : l’émission de l’ordonnance par le prescripteur
Cette étape, est celle de l’utilisation par le médecin pour rédiger sa prescription et le pharmacien pour la dispenser, des moyens informationnels mis à sa disposition par les LAPs ou les LADs. Historiquement, les LAPs sont apparus avant les BdMs et les médecins ont constitué alors, manuellement, les outils nécessaires à l’édition des ordonnances : fichier médicaments restreint à quelques centaines de médicaments et éventuellement thesaurus de mots pour les antécédents, les signes et les pathologies. Certains LAPs proposaient et proposent encore des formulaires simplifiés de rédaction de la posologie. Lorsque les BdMs ont commencé à être intégrées dans les LAPs, d’abord pour un fichier spécialités exhaustif, les éditeurs de LAPs ont développé des outils de codage des pathologies issus des BdMs. Mais l’expérience montre que, dans leur grande majorité, les médecins, en particulier en libéral, enregistrent encore dans leurs dossiers patient leurs observations en verbatim, simple transposition électronique du dossier papier, et qu’une minorité d’entre eux utilise des nomenclatures diverses : CIM.10, CISP , DRC , thesaurus divers proposés par des tiers tels que les éditeurs de LAP, voire un thesaurus personnel constitué au fil des consultations éventuellement mutualisé avec d’autres utilisateurs par le biais des éditeurs.
En conséquence, pour l’essentiel, les alertes générées dans ces conditions par les LAPs concernent uniquement les données issues des spécialités ne nécessitant pas ou peu un croisement avec les données du Dossier Patient Informatisé (DPI). Ces fonctions concernent surtout le choix du médicament, les interactions médicamenteuses, les posologies, les effets indésirables, le risque pour la conduite automobile. Pour la sécurisation complète de la prescription, le prescripteur s’appuie alors sur son expérience personnelle, la consultation du DPI et éventuellement la lecture des monographies textuelles des spécialités. La situation est assez similaire en milieu hospitalier et l’observation de nombreux LAPs hospitaliers a montré qu’exception faite des interactions, les fonctions de prévention de la iatrogénie demeurent restreintes.
Les LADs, au contraire, ont été dès leur apparition appariés avec des BdMs fournissant un fichier produit exhaustif nécessaire à la facturation et un outil de codage des grandes pathologies et des données staturo-pondérales avec possibilité restreinte d’enregistrer du verbatim. Conséquence logique de cet encadrement assez rigoureux, les LADs génèrent outre la détection des interactions et des posologies maximales usuelles, des alertes tenant compte des dossiers patients (pathologies, âge, sexe, grossesse, etc.).
En résumé, l’analyse des failles du processus informationnel sur le médicament peut s’exercer sur trois points critiques :
- La transformation de l’information primaire en données structurées par les BdM ;
- L’intégration des BdMs dans les LAPs et les LADs ;
- La réalité du codage du Dossier Patient Informatisé (DPI) par les prescripteurs.
Les modes de défaillance et leur criticité
Les défaillances effectivement constatées ont été classées par catégories et, pour chaque catégorie, répartis en trois degrés de RPN.
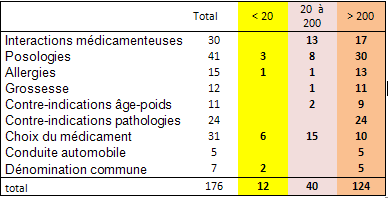
Sur 176 défaillances constatées :
70 % ont un score RPN > 200
23 % ont un score RPN compris entre 20 et 200
7 % ont un score RPN < 20
Défaillances dont le score RPN est > 200
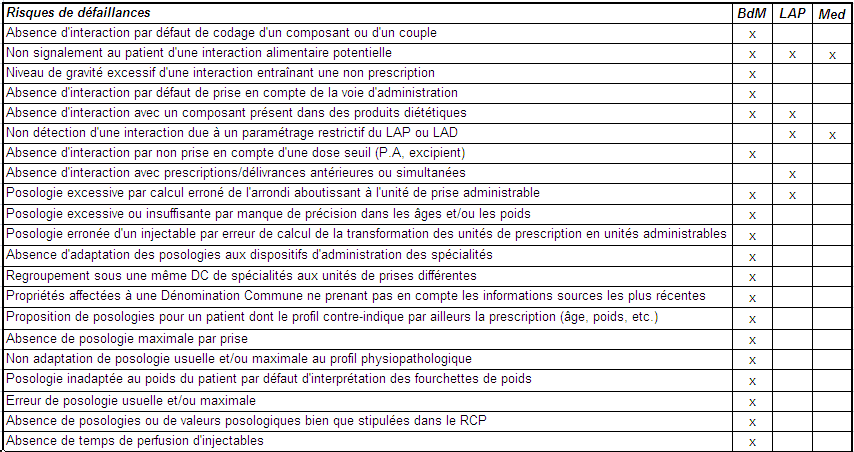
Sur les défaillances dont les scores RPN sont > 200
71% peuvent être attribuées aux BdMs
21% peuvent être attribuées aux LAP
8% peuvent être attribuées aux prescripteurs
Les posologies
De nombreux facteurs peuvent générer une posologie inadaptée.
Certains LAPs, qu’ils soient de médecine ambulatoire ou hospitaliers ne proposent pas de posologies préconstruites à partir du RCP. Cette sobriété sur ce point crucial contribue probablement à un risque pointé dans la littérature comme majeur.
La certification des LAPs n’exige pas la mise à disposition de posologies structurées, se limitant à la nécessité au contrôle du dépassement des posologies maximales par 24 heures et en durée de traitement, sans prise en compte des variations liées aux indications, à l’âge, au poids, aux traitements associés, à la grossesse, au degré d’insuffisance rénale ou hépatique, etc. La conséquence de la non structuration des posologies est que ces dernières ne peuvent pas prendre en compte les données staturo-pondérales ou physiopathologiques et qu’il appartient au prescripteur consultant le texte de remplir le formulaire de posologie proposé par le LAP en choisissant hors de toute aide les doses, les unités d’administration voire la forme galénique, les éventuelles dilutions à réaliser, les séquences de prises, la durée du traitement, les modes d’administration….
Dans le cas où sont proposées des posologies structurées, il est toutefois possible de les utiliser sans croisement avec le dossier patient, en choisissant dans une liste la posologie adéquate au patient. Les risques, que le dossier patient soit codé ou non, sont principalement la non-conformité aux données du RCP (chapitre posologie mais également d’autres chapitres où des restrictions posologiques existent comme les indications, contre-indications, précautions d’emploi, interactions, grossesse, pharmacocinétique, instructions pour l’utilisation et la manipulation, dispositif d’administration, voire conservation), l’absence ou l’erreur de valeur posologique, des posologies inadaptées au profil physiopathologique du patient (âge, sexe, poids, taille, degré d’insuffisance rénale ou hépatique), la non adaptation à la présentation et/ou au dosage, etc.
Ont été notamment repérés :
- Des posologies ne prenant pas en compte ou de manière erronée le profil du patient (âge, poids, taille, insuffisance rénale, insuffisance hépatique, grossesse, etc.)

- Des erreurs ou des absences de doses usuelles ou maximales par prise ou par jour

- Des posologies non exprimées en unités de prise administrables compréhensibles par l’infirmier ou le patient (ex : conversions d’unités de masse mg, g, mmoles, en cuillère-mesure, gouttes, kg-graduation, ml de solution injectable à différentes concentrations, ml de seringue doseuse buvable, etc.).

- La dose calculée en fonction du poids ou de la surface corporelle du patient est nettement inférieure à l’unité de prise administrable (comprimé, ampoule, flacon, stylo, etc.) mais la BdM l’arrondit à même unité de prise exposant alors au risque de surdosage (erreur médicamenteuse relevée par la cellule erreurs médicamenteuses de l’ANSM).
- Des posologies non adaptées aux différentes présentations d’une même spécialité
Les interactions médicamenteuses
Fonction historique des LAPs et des LADs, la détection des interactions médicamenteuses implique les composants des spécialités de l’ensemble de la prescription et leur voie d’administration sans nécessiter l’instanciation du dossier patient.
Les sources de l’information sur les interactions sont le « Thesaurus des interactions médicamenteuses » de l’ANSM et à défaut le chapitre « Interactions » des RCP de l’ANSM et de l’EMA. Les interactions sont classiquement hiérarchisées en 4 niveaux de gravité (hormis dans les AMM européennes).
Le niveau de gravité de l’interaction peut dépendre de l’indication. L’interaction doit alors être différenciée au niveau de la spécialité.
Le premier risque est la non détection d’une interaction médicamenteuse potentielle dont les causes sont multiples :
- Non respect du Thesaurus des Interactions médicamenteuses de l’ANSM ;

- Non prise en compte d’un composant dans une classe d’interactions ;
- Non prise en compte d’une dose seuil d’un principe actif, voire d’un excipient ;
- Non prise en compte d’un couple d’interactions ;
- Non respect d’un niveau de gravité d’une interaction

- Absence de composant dans le thésaurus des interactions ;
Le Thésaurus des Interactions Médicamenteuses de l’ANSM étant mis à jour 1 à 2 fois par an, certains composants de spécialités nouvelles peuvent ne pas y être référencés. Si les informations du RCP ne sont pas utilisées pour les spécialités nouvelles dont les composants ne sont pas mentionnés dans le thesaurus, l’absence d’alerte peut engendrer une fausse sécurité de la prescription et être source d’effets indésirables.
L’absence d’alerte ou une alerte d’un niveau de gravité inférieur à celui précisé dans la source est potentiellement génératrice d’effets indésirables pouvant nécessiter une hospitalisation (voire une IVG en cas de grossesse non désirée par inefficacité d’un traitement contraceptif). Cette ordonnance est ensuite vérifiée par le dispensateur, susceptible de détecter l’erreur sous réserve que le système d’alerte disponible dans le LAD lui signale. Si ce dernier s’appuie sur la même BdM que celle du prescripteur, l’erreur de ne sera pas détectée sauf si le dispensateur se réfère aux sources primaires.
Lorsque le logiciel alerte le professionnel de santé avec un niveau de gravité supérieur à celui précisé dans la source, la conséquence peut en être la non prescription d’un médicament pouvant être nécessaire au traitement du patient.
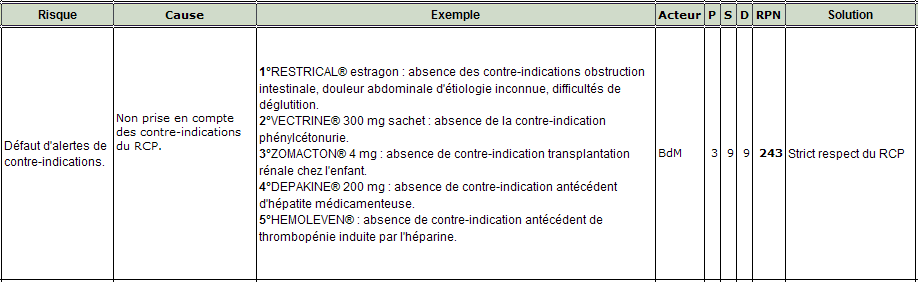
Les contre-indications par rapport aux données du patient
A la différence des interactions médicamenteuses et des posologies, la détection des contre-indications potentielles d’une ligne de prescription implique un croisement entre les informations sur la spécialité issues de la BdM et les données du patient issues du DPI. Le non signalement d’une contre-indication est une défaillance majeure sur plusieurs points :
Les allergies dont la gestion est variable d’une BdM à l’autre peut être rendue complexe par l’existence d’allergies croisées et par la nature des substances susceptibles d’être allergènes (principes actifs et/ou excipients et/ou résidus du procédé de fabrication). L’absence d’alertes peut être à l’origine de réactions allergiques potentiellement graves (Œdème de Quincke, choc anaphylactique) voire létales.

La gestion des allergies suppose que soient pris en compte les excipients, certains d’entre eux étant impliqués dans les réactions d’allergies croisées (colorants azoïques par exemple).

L’âge du patient est souvent présent dans le chapitre contre-indications, voire précautions d’emploi comme un facteur de risque mais l’expression des fourchettes d’âge, très diversifiée, impose une gestion précise des algorithmes de décodage ne pouvant s’appuyer sur les tranches classiques nourrisson, enfant, adulte.

Le poids peut être un paramètre plus important que celui de l’âge (ex : pour le PARACETAMOL 1 g dont l’utilisation est restreinte à l’enfant de plus de 50 kg même s’il a moins de 15 ans et non à l’enfant de plus de 15 ans).
La grossesse de même que l’allaitement ne sont pas toujours mentionnés directement dans le chapitre contre-indication du RCP et cependant la lecture du chapitre grossesse et allaitement permet de lui affecter ce niveau de gravité (contre-indiqué ou déconseillé). La pratique courante des utilisateurs de LAPs et LADs, de rendre inopérantes les alertes de précaution d’emploi, ajoute au risque de non détection d’un risque pendant la grossesse ou l’allaitement.

La grossesse, d’expression variable d’une spécialité à l’autre, ne peut se satisfaire de l’expression classique en trimestres mais doit être précisée en semaines d’aménorrhée ou de grossesse, sachant en outre que la gravité des contre-indications d’une spécialité peut varier selon les différentes étapes de la grossesse.
Dans certains cas, la traduction des semaines de grossesse en trimestres peut générer des paradoxes. Si un médicament est contre-indiqué à partir du 6ème mois, sa transformation en troisième trimestre (soit à partir du début du 7ème mois) laisse un mois sans contre-indication.

Compte-tenu des risques liés à certains médicaments concernant la conduite de véhicules et de machines (accidents de travail ou de véhicules avec leurs conséquences en matière de handicaps ou de décès), le médecin et ou le pharmacien sont tenus d’en informer le patient. Leur logiciels doivent en conséquence les alerter sur ce danger potentiel.
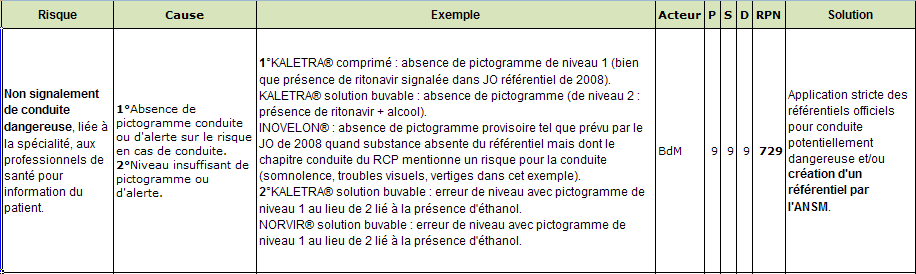
Les pathologies sont fréquemment un risque aggravé de contre-indications signalé dans les sources. L’efficacité de la génération des alertes dépend alors étroitement de la fidélité du codage des pathologies par la BdM et de leur croisement avec la méthode d’enregistrement des pathologies dans le DPI par le prescripteur.
Lorsque des spécialités sont présentes sans aucune information pharmaco-clinique, l’absence d’alertes qui en est la conséquence est une défaillance dont le risque peut être élevé.
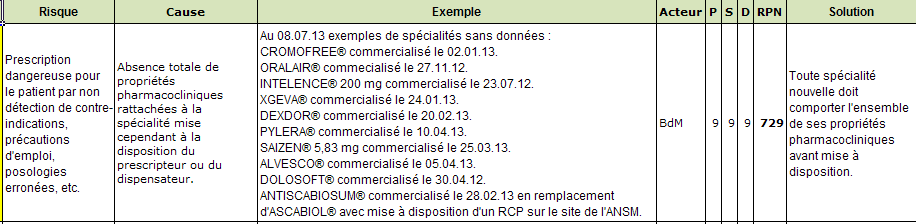
Si le DPI est peu ou pas renseigné de manière structurée par le professionnel de santé, les fonctions de génération d’alerte proposées par la BdM seront inactives. Dans le cas contraire, le croisement efficace entre données médicaments et données patient suppose que plusieurs conditions soient remplies :
- L’interopérabilité des nomenclatures utilisées par le codage des spécialités et du dossier patient ;
- L’efficacité des algorithmes de gestion des alertes ;
- L’intégration correcte des fonctions d’alerte fournies par les BdMs dans les logiciels mis à disposition des professionnels de santé ;
- Le respect des contre-indications du RCP.
Les erreurs de choix du médicament
Les auteurs signalent comme fréquentes les erreurs provenant de la prescription erronée d’un médicament autre que celui que le prescripteur avait décidé de prescrire (dérapage de souris, choix du mauvais dosage, etc.).
Les risques d’erreurs liés au choix du médicament sont de deux types : erreur dans la sélection du médicament recherché ou inadaptation du médicament sélectionné compte-tenu des caractéristiques du patient.
Les critères de recherche d’une spécialité ou d’une présentation sont divers et présentés dans le tableau d’analyse. En fonction des critères de recherche choisis par l’utilisateur, le logiciel d’aide propose une liste restreinte de spécialités.
L’objectif de la recherche est soit de prescrire ou délivrer le médicament recherché, soit d’obtenir des informations sur les propriétés de ce médicament.
Deux risques possibles :
Liste incomplète ou vide.
L’utilisateur peut cependant prescrire ou délivrer en créant dans son logiciel le seul libellé de la spécialité. Les propriétés pharmaco-cliniques étant alors vides, le risque est lié à l’absence d’alertes en matière d’interactions médicamenteuses ou de contre-indications. L’utilisateur peut également s’abstenir de prescrire, privant ainsi le patient d’un médicament nécessaire au traitement de son état et peut-être sans alternative thérapeutique.

Résultat erroné.
La liste résultant de la recherche comporte des spécialités ne répondant pas au critère de recherche. Le risque est alors celui de prescrire un médicament ne répondant pas au besoin du patient, risque qui apparaît comme une des premières causes des effets indésirables des médicaments.
La prescription en dénomination commune
Rendue obligatoire par la certification des LAPs, la prescription en dénomination commune introduit des risques spécifiques à cette méthode. Elle nécessite l’élaboration de médicaments virtuels comportant le libellé du ou des principes actifs, leur(s) dosage(s), la forme pharmaceutique, éventuellement la ou les voies d’administrations lorsque nécessaires à l’identification précise (ex. amoxicilline 1 g IM ou amoxicilline 1 g IM/IV), et surtout des propriétés pharmaco-cliniques et des liens vers les spécialités susceptibles d’être délivrées par le pharmacien lors de l’exécution de la prescription.
Un exemple de défaillance rencontrée parmi d’autres :

Le médecin a sélectionné, pour un enfant de 30 kilos, la spécialité ADVIL® 20 mg/ml solution buvable (dosée à 7,5 mg d’ibuprofène par kg/graduation) et choisi la posologie proposée de 30 kg/graduation 4 fois par jour soit au total 900 mg d’ibuprofène/jour puis fait traduire le libellé d’ADVIL® par son logiciel en DC. La prescription devient : IBUPROFENE 20 mg/ml solution buvable avec la posologie ci-dessus définie.
A la lecture de l’ordonnance en DC, le pharmacien choisit de délivrer NUROFENPRO® 20 mg/ml solution buvable (dosé à 10 mg d’ibuprofène par kg/graduation). Mais avec la posologie telle que rédigée par le médecin, la dose totale journalière est alors de 1200 mg d’ibuprofène. Le risque de surdosage est réel si la posologie rédigée est appliquée.
Le pharmacien aurait pu constater le problème et délivrer ADVIL® (NUROFENPRO® étant limité à 3 prises par jour), mais la seule solution sécurisée est que l’ANSM modifie le libellé des spécialités en cause : ADVIL® 20 mg/ml (7.5 mg/kg graduation), solution buvable et NUROFENPRO® 20 mg/ml (10 mg/kg graduation) solution buvable et révise également les libellés dans les cas analogues pour que le dosage des spécialités soit directement exprimé ou complété par la teneur en unité de prise administrable.
Les risques liés à la prescription en dénomination commune peuvent être situés à plusieurs niveaux mais l’absence d’interopérabilité est sans doute le risque majeur. En effet, en l’absence d’un référentiel de la prescription en dénomination commune sous forme d’une liste de médicaments virtuels (avec un lien vers les spécialités correspondantes), chaque BdM a construit son propre référentiel avec une codification propriétaire. En conséquence, l’interopérabilité entre les LAPs et les LADs n’est pas assurée et impose une transmission de ce mode de transmission en verbatim sans garantie d’identification non ambigüe.
A la veille de la dématérialisation de la prescription pour assurer la transmission électronique des prescriptions entre professionnels de santé, l’absence d’interopérabilité des prescriptions en dénomination commune ajoutée à l’obligation d’utiliser cette méthode de prescription prévue par le législateur est un des points critiques.
Il existe un second risque lié à la prescription en dénomination commune lié à une gestion défaillante des contre-indications :
- Si la BdM utilise les propriétés pharmaco-cliniques d’une des spécialités pour les affecter à la spécialité virtuelle, des spécificités propres à cette spécialité peuvent altérer la fiabilité des alertes (ex : alertes liées aux excipients à effet notoire propre à chaque spécialité alors qu’ils sont à exclure de la spécialité virtuelle, paracétamol sans prise en compte de la dose maximale de 3 g par jour chez l’adulte de moins de 50 kg, posologies non généralisables comme dans le cas des suspensions pédiatriques de paracétamol, des théophyllines, des patchs d’antalgiques, etc.) ;
- Si la BdM ne met pas à jour les informations du médicament virtuel suite à la révision des spécialités rattachées. Au même titre que les spécialités commercialisées, la fraicheur de l’information pharmaco-clinique du médicament virtuel est un élément de la sécurisation de la prescription.
Les défaillances à l’étape du prescripteur et du dispensateur
Autant il est possible de repérer les défaillances à l’étape des BdMs et des LAPs et des LADs, autant l’étape des prescriptions c’est-à-dire des ordonnances demeure actuellement peu explorée.
En ville, les pharmaciens scannent les ordonnances puis les transmettent à l’assurance maladie mais ces données sont sous format verbatim. Ceci est d’autant plus regrettable qu’à ce niveau les défaillances éventuelles sont devenues de réelles erreurs.
Il a été impossible d’accéder à un volume significatif ordonnances réelles prescrites par des médecins, remises aux patients et dispensées par des pharmaciens qui auraient permis de vérifier si les défaillances constatées dans la chaîne de l’information devenaient des erreurs médicamenteuses proprement dites.
Suggestion de solutions correctives ou préventives
Concernant les scores les plus élevés de défaillances, nous proposons des recommandations-solutions susceptibles de pallier les défaillances constatées.
Concernant d’une manière générale la qualité des BdMs un contrôle systématique indépendant de leur qualité s’inspirant des travaux de Coste et coll. (14) permettrait d’informer les éditeurs des défaillances constatées afin d’y remédier et d’éviter qu’elles ne se retrouvent dans les LAPs et les LADs.
Interactions médicamenteuses
Recommandation de la création et mise à jour par l’ANSM, auteur du thésaurus des interactions médicamenteuses, d’un dossier numérique structuré qui en soit la représentation et qui soit directement intégrable par les BdMs et/ou les LAPs et LADs. Ceci implique la fourniture de tables référentielles liées : composants, voies d’administration, voire indications et ou spécialités concernées par un couple (lorsque l’interaction dépend de l’indication, d’une dose seuil, de la présence d’un salifiant en quantité notable, etc.).
Contre-indications
Allergies
Fourniture par l’ANSM d’un référentiel structuré des composants (principes actifs, excipients et résidus de procédés de fabrication) incluant les liens d’allergies croisées entre composants.
Pathologies
- Fourniture par l’ANSM d’un thesaurus référentiel des spécialités proche du langage naturel comportant en attributs les codes CIM.10 d’indications, de contre-indications et de précautions d’emploi.
- Obtenir des BdMs que toute spécialité référencée mise à disposition des LAPs et LADs soit renseignée avec l’ensemble de ses propriétés pharmaco-cliniques.
Grossesse et allaitement
- Normalisation du stade de grossesse applicable par les BdMs, les LAPs et les LADs (semaines de grossesse ou d’aménorrhée).
- Unification des informations grossesse et allaitement dans les RCP (dans le chapitre grossesse et avec un niveau de gravité ne nécessitant pas d’interprétation).
Age et/ou poids du patient
Normaliser l’expression des tranches d’âge et de poids dans les spécialités définissant sans ambigüité l’interprétation d’un âge ou de poids compris entre « de » et « à »
Choix des médicaments
- Fourniture par l’ANSM, pour toute spécialité, d’un nom court compatible avec les possibilités d’affichage des LAPs et LADs (environ 40 caractères).
- Classer les spécialités de telle manière que les divers dosages d’une même spécialité soient affichés par dosages croissants.
- Création par l’ANSM d’un référentiel exhaustif des composants (principes actifs, excipients et synonymes).
- Raccourcissement du délai de mise à disposition des spécialités nouvelles par les BdMs, les LAPs et les LADs (dès commercialisation).
- Modification par l’ANSM de certains libellés de spécialités les dosages spécifiés en unités de prises administrables.
Posologies
Lorsque les posologies sont exprimées dans le RCP en unités de masse, de masse/volume, de pourcentage voire de millimoles, fournir les ratios mathématiques permettant leur traduction exacte en unités de prises administrables (ANSM, BdMs).
Prescription en dénomination commune
- Fourniture par l’ANSM d’un référentiel des médicaments en Dénomination Commune et de leurs liens vers les spécialités.
- Respecter les restrictions de la prescription en DC préconisées par la HAS et les RCP.
Non codage du DPI par le professionnel de santé
La correction du faible niveau de codage du DPI est un chantier important qui conditionne fortement la prévention de la iatrogénie.
Plusieurs solutions non exclusives peuvent être envisagées :
- Fourniture par l’ANSM d’un thesaurus référentiel proche du langage naturel comportant en attributs les codes CIM.10 d’indications, de contre-indications et de précautions d’emploi.
- Mise en œuvre par les LAPs et les LADs du rétro-codage des pathologies à partir des indications des spécialités prescrites ou délivrées.
- Normalisation des méthodes de codage de la grossesse, de l’âge et autres valeurs physiopathologiques permettant l’interopérabilité entre les informations des BdMs et celles enregistrées dans le DPI.
- Analyse de lots d’ordonnances et repérage des défaillances. Cette étude serait seule susceptible d’identifier et d’évaluer les erreurs les plus fréquentes et réelles.
Discussion
L’étude n’est pas exhaustive et ne prétend pas avoir balayé la totalité des erreurs constatées, seules les plus significatives ont été relevées. Les résultats des observations ne sont pas des erreurs médicamenteuses au vrai sens du terme car n’ayant pas donné lieu à des administrations à des patients mais des risques de défaillance.
L’étude a été circonscrite aux risques d’erreurs les plus fréquemment cités dans la littérature : erreurs de médicaments, posologies, interactions médicamenteuses, contre-indications par rapport au profil du patient (allergies, données physiopathologiques), grossesse et allaitement. La prescription en Dénomination Commune, compte-tenu de sa récente montée en charge, a fait l’objet d’une série de tests spécifiques.
Afin de concentrer les tests sur les risques majeurs, ont été exclus de l’étude des risques d’erreurs qui mériteraient cependant une analyse analogue : incidence des excipients à effet notoire, respect du G6PD, suivi des effets indésirables, conformité des monographies aux textes officiels, liens avec les documents officiels de l’ANSM ou de la HAS, influence des voies d’administration, séquençage des posologies, etc.
Enfin, ont été délibérément exclus les risques n’étant pas à proprement parler des risques iatrogènes tels que les prix, les génériques, le SMR et l’ASMR, les conditions de délivrance et de prescription, les taux de remboursement (selon les indications si tel est le cas), les coûts de traitement.
Les sources primaires référentielles utilisées ont été principalement le Thésaurus des Interactions Médicamenteuses de l’ANSM, les RCP des spécialités disponibles sur les sites de l’ANSM et de l’EMA, le Guideline européen des excipients à effet notoire, le JO (pictogramme conduite, dopant).
Comme sources secondaires, nous avons utilisé, entre mai et juillet 2013 les sites Internet des BdMs THESORIMED et THERIAQUE ainsi que les logiciels autonomes (standalone) des BdMs VIDAL (Vidal Expert) et CLAUDE BERNARD (BCB autonome), toutes les 4 agrées par la HAS. Les spécialités prises en exemple sont des spécialités commercialisées. Les résultats des tests ont été systématiquement comparés aux sources primaires correspondantes.
Le propos n’était pas de comparer les BdMs, les LAPs ou les LADs entre eux.
Les tests n’ont pas vocation à l’exhaustivité, ils ont été choisis parmi de nombreuses constatations car représentatifs des erreurs rencontrées. Nous avons vérifié leur permanence dans un LAP ou un LAD. Il s’avère expérimentalement qu’une erreur présente dans une BdM n’est pas corrigée dans les LAPs et les LADs utilisant cette BdM.
La constatation d’une défaillance dans notre étude ne préjuge pas de son devenir dans la vraie vie, le médecin comme le pharmacien pouvant naturellement exercer leur expertise et leur intelligence comme moyens de contrôle avant que le risque d’erreur ne devienne une erreur réelle mais il apparaît cependant indispensable que l’information dont ils disposent dans leurs logiciels n’induise pas d’erreurs.
Conclusion
Cette étude permet de constater l’existence de défaillances dans la chaine de transmission et de transformation de l’information sur le médicament depuis les sources primaires opposables jusqu’à la rédaction de l’ordonnance par le médecin et sa dispensation par le pharmacien. La phase critique étant celle de la transformation de l’information source verbatim en information structurée par les BdMs.
Des solutions correctives sont proposées pour chaque défaillance constatée concernant les BdMs, les LAPs et les LADs mais la proposition principale est que l’ANSM, fournisseur légitime de l’information source, en fournisse également les éléments structurés afin d’en garantir la conformité et d’assurer l’interopérabilité entre les données patients et les données sur le médicament.
Une proposition générale pour pallier les défaillances éventuelles de la structuration de l’information serait de mettre en place un organe indépendant de contrôle des BdMs utilisant des indicateurs quantitatifs permettant d’en mesurer la qualité.
Les ordonnances remises aux patients demeurent un territoire presque inexploré en matière d’erreurs médicamenteuses exception faite des observations des pharmaciens hospitaliers pratiquant l’analyse pharmaceutique et les signalements adressés par les professionnels de Santé à la cellule erreurs médicamenteuses de l’ANSM. Une évaluation approfondie de cette dernière étape du cheminement de l’information permettrait de vérifier si les défaillances constatées se transforment en erreurs médicamenteuses proprement dites.
Références
- Latil F. Place de l’erreur médicale dans le système de soins. Acta Endosc. 2007 Aug 1;37(4):509–20.
- Kohn LT, Corrigan JM, Donaldson MS. To Err Is Human: Building a Safer Health System. [Internet]. 2002 [cited 2013 Jul 14]. Available from: http://informahealthcare.com/doi/pdf/10.1080/1356182021000008364
- European Medicines Agency. Medication-errors workshop [Internet]. London, UK; 2013 Mar. Report No.: EMA/144458/2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Report/2013/05/WC500143163.pdf
- Cousins DH, Gerrett D, Warner B. A review of medication incidents reported to the National Reporting and Learning System in England and Wales over 6 years (2005-2010). Br J Clin Pharmacol. 2012 Oct;74(4):597–604.
- Agence française de sécurité sanitaire des produits de santé. Guichet erreurs médicamenteuses: Présentation et bilan depuis la mise en place. Saint Denis, France; 2009 Juin.
- Wolfstadt JI, Md JHG, DSc TSF, RPh ML, Sunila Kalkar MBBS MD, MSc WW, et al. The Effect of Computerized Physician Order Entry with Clinical Decision Support on the Rates of Adverse Drug Events: A Systematic Review. J GEN INTERN MED. 2008 Apr 1;23(4):451–8.
- Ammenwerth E, Schnell-Inderst P, Machan C, Siebert U. The effect of electronic prescribing on medication errors and adverse drug events: a systematic review. J Am Med Inform Assoc. 2008 Oct;15(5):585–600.
- Berger RG, Kichak JP. Computerized physician order entry: helpful or harmful? J Am Med Inform Assoc. 2004 Apr;11(2):100–3.
- Liot P, Toussi M, Anglade I, Simon G, Nabarette H. Designing explicit criteira to control the quality of outpatient electronic prescribing systems in France. Quality and Safety in Health Care. 2008 Aug;17(4):14–5.
- Hacin L, Mainar A, Édouard B. Évaluation des bases de données médicamenteuses disponibles sur le marché français. Annales Pharmaceutiques Françaises. 2013 Mar;71(2):123–34.
- DeRosier J, Stalhandske E, Bagian JP, Nudell T. Using health care Failure Mode and Effect Analysis: the VA National Center for Patient Safety’s prospective risk analysis system. Jt Comm J Qual Improv. 2002 May;28(5):248–67, 209.
- Lago P, Bizzarri G, Scalzotto F, Parpaiola A, Amigoni A, Putoto G, et al. Use of FMEA analysis to reduce risk of errors in prescribing and administering drugs in paediatric wards: a quality improvement report. BMJ Open. 2012;2(6).
- Venot A, Burgun A. Informatique médicale, e-santé: fondements et applications. Paris; Berlin; Heidelberg [etc.]: Springer; 2012.
- Coste J., Séné B., Milstein C., Bouée S., Venot A., “ Indicators for the automated analysis of drug prescribing quality”, Methods of Information in Medicine, 37 (1998): 38-44.
Partager cette page :

- Précédent
- Suivant